










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortugaliae Electrochimica Acta
Print version ISSN 0872-1904
Port. Electrochim. Acta vol.29 no.5 Coimbra Sept. 2011
https://doi.org/10.4152/pea.201105309
Electron Reservoir Activity of High-Nuclearity Transition Metal Carbonyl Clusters
Plenary lecture at the XII Iberic Meeting of Electrochemistry and XVI Meeting of the Portuguese Electrochemical Society
Fulvio Rossi and Piero Zanello*
Dipartimento di Chimica dell' Università di Siena, Via A. De Gasperi 2, 53100 Siena, Italy
doi: 10.4152/pea.201105309
Abstract
Metal carbonyl clusters are molecules or molecular ions perfectly defined in size, composition and structural details, which belong by size to the field of nanomaterials. Their molecular structures result from subtle balances between the metal-metal and metal-carbonyl interactions and usually adopt close-packed structures in which a chunk of cubic or hexagonal metal lattice is surrounded by a shell of CO ligands. Very often, such derivatives display extended redox activity affording reversible electron cascades. In many cases such activity increases if interstitial or semi-interstitial atoms of the main group elements (C, N, P, etc.) are inserted in their frames. This in fact triggers establishment of further metal-to-interstitial atom(s) interactions which not only contribute to the number of cluster valence electrons, but also modifies the bonding character of the frontier molecular orbitals.
Keywords: high-nuclearity metal-carbonyl clusters, molecular structures, electrochemistry.
Introduction
Miniaturization constitutes an actual and future challenge in developing mechanical, optical, and electronic devices. Metal clusters having a metallic core encapsulated in a ligand shell could be suitable candidates as components of data storage devices and could potentially represent the ultimate solution for miniaturization in nanoelectronics [1].
As a matter of fact, metal-carbonyl clusters can be considered in principle as microscopic capacitors in that they are constituted by a kernel of metal atoms surrounded by a shell of nominally insulating carbonyl ligands, Fig. 1 [1a].
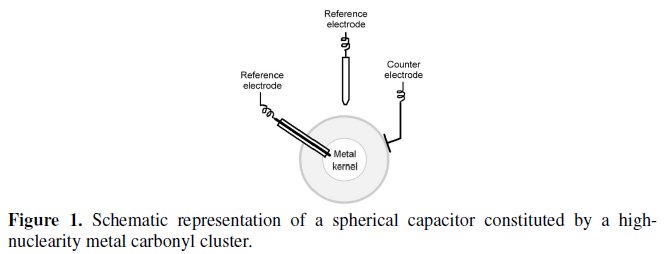
Nevertheless, such a morphological similarity must be accompanied by a series of further requirements, such as:
1. electron-sink behavior, i.e. they should be able to accept and release electrons reversibly (or they must maintain unaltered their original molecular structure);
2. the carbonyl ligand shell should effectively insulate the metallic core in order to prevent intermolecular exchange of electrons, which would hinder effective storage of electrons in the core. In fact, easy intermolecular electron exchanges prevent the effective storage of electrons in the molecule. In this connection, evidences of the ability of metal cores of high-nuclearity metal carbonyl clusters to act as a quantum dot have been gained;
3. the metallic core of the cluster should possibly undergo transition from insulator-to-semiconductor regime;
4. finally, for practical purposes, the metallic core should display dimensions falling in the nanometric field.
The prerequisite to act as capacitor is to possess an extended and reversible redox behavior.
Interstitial or semi-interstitial atoms of the main group elements (C, N, P, etc.) play an essential role in the stabilization of high-nuclearity metal carbonyl clusters not only because they can contribute to the number of cluster valence electrons with no sterical consequences on the geometry of the cluster surface, but also because they can modify the bonding character of the frontier molecular orbitals [2].
In this picture, we wish to give an overall survey of the redox propensity of a considerable number of metal-carbonyl clusters as it arises from electrochemical investigation [3].
Homometallic clusters
Cobalt clusters
Let us start with the 130-CVE (Cluster Valence Electrons) dianion [Co9(μ8P)(CO)21]2-. Fig. 2 shows its monocapped square antiprismatic geometry (encapsulating the phosphide atom) as well as its redox activity in MeCN solution [4].
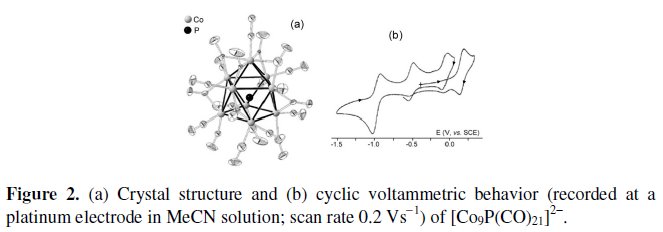
It exhibits a one-electron oxidation and a two-electron reduction, both of which are partially chemically reversible. In addition, both the redox processes are coupled to chemical complications which afford two new peak systems at intermediate potential values, which have been tentatively assigned to degradation of the electrogenerated species to the phosphide monoanion [Co6P(CO)16]-.
Related to the previous dianion is the 142-CVE bicapped square antiprismatic phosphide trianion [Co10(μ8-P)(CO)22]3-, whose molecular structure and electrochemical behavior are illustrated in Fig. 3 [4].

As seen, it exhibits a redox pattern somewhat reminiscent that of [Co9P(CO)21]2-, in that it undergoes a one- electron oxidation and two sufficiently separated one-electron reduction processes. All the processes exhibit partial chemical reversibility. The formal electrode potentials of the redox changes exhibited by the two phosphide clusters are compiled in Table 1, together with those of related species that will be discussed below.
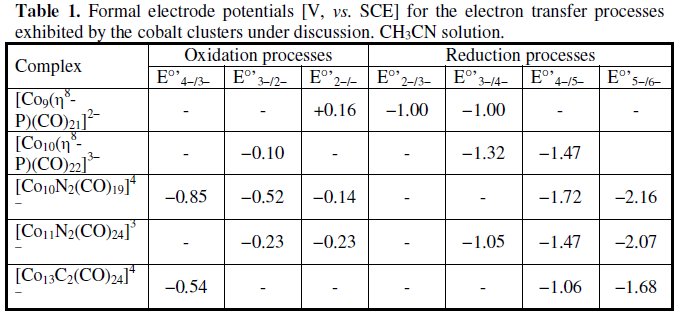
In confirmation that cobalt clusters with interstitial or semi-interstitial nitrogen atoms show a distinctive aptitude towards unusual stereochemistry and high coordination numbers, (i.e. affording quite spectacular shapes and extended electron transfer properties), let us consider the anions [Co10N2(CO)19]4- and [Co11N2(CO)21]3-.
Fig. 4 shows the crystal structure [5a] and the cyclic voltammetric behavior in CH3CN solution [5b] of the 142-CVE tetraanion [Co10N2(CO)19]4-.
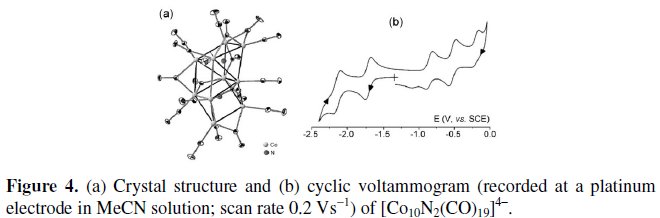
The tetraanion (whose molecular structure can be seen as built from two trigonal prismatic units each containing an interstitial nitrogen atom, sharing a common edge) undergoes either three successive oxidations or two successive reductions, which, but for the most anodic step, exhibit features of chemical reversibility in the cyclic voltammetric time scale.
In confirmation of the complete chemical reversibility of such processes, cyclic voltammetric tests carried out on the solutions resulting from each exhaustive one-electron removal afforded profiles quite complementary to the original ones.
Given the similar peak heights of the reduction processes, it follows that the present cluster can shuttle reversibly the rich sequence [Co10N2(CO)19]2-/3-/4-/5-/6-.
In turn, Fig. 5 shows the molecular structure and the voltammetric profile in CH3CN solution of the 154-CVE trianion [Co11N2(CO)21]3- [5b].
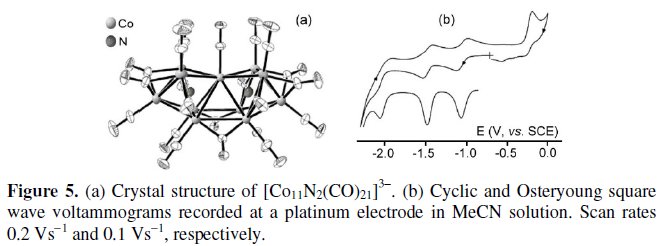
The trianion (whose molecular structure can be seen as two octahedrons having a common vertex, each octahedron encapsulating a nitrogen atom) exhibits three consecutive reductions and one oxidation process, each step involving one electron per molecule. The pertinent formal electrode potentials are summarized in Table 1.
The last example of the present section we discuss is the 177-CVE tridecacobaltate dicarbide tetranion [Co13C2(CO)24]4-. Its crystal structure can be described as two mutually rotated prismatic Co6C units sharing a common vertex, capped at opposite sides by two five-bridging cobalt atoms, Fig. 6 [6a].
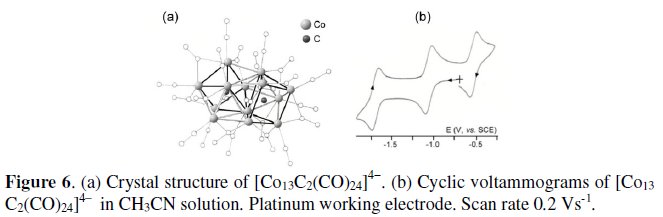
As illustrated in Fig. 6b, the tetraanion exhibits one oxidation and two reduction processes, all having features of chemical and electrochemical reversibility. Each passage involves one electron [6b]. The formal electrode potentials of such redox changes are reported in Table 1.
We note that the related 176-CVE dinitride trianion [Co13N2(CO)24]3- possesses a more limited redox activity [6b].
Iridium clusters
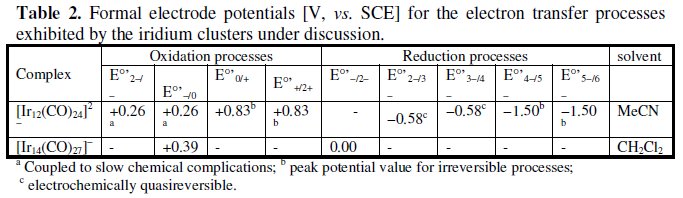
Two examples of iridium carbonyl cluster with quite spectacular molecular structures are here reported. The first example is constituted by the 158-CVE dianion [Ir12(CO)24]2-, whose metallic frame can be simply described as a large (n2) incomplete trigonal bipyramid, in that it lacks one axial and one equatorial vertex, Fig. 7a [7]. In MeCN solution it undergoes a series of oxidations and reductions coupled to chemical complications. The most significant are the first two-electron oxidations and the first two-electron reductions illustrated in Fig. 7b [7].
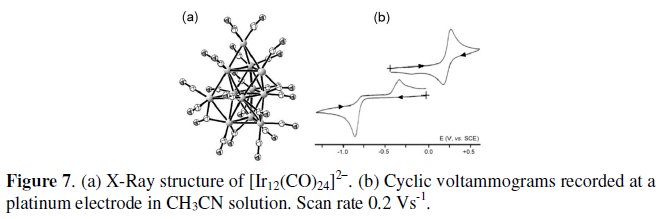
In spite of features of chemical reversibility of the [Ir12(CO)24]2-/[Ir12(CO)24]0 oxidation, the electrogenerated neutral derivative suffers slow decomposition. As far as the [Ir12(CO)24]2-/[Ir12(CO)24]4- reduction is concerned, although largely departing from the electrochemical reversibility (the high peak-to-peak separation attests its electrochemical quasireversibility), it resulted chemically reversible upon exhaustive electrolysis, thus supporting that the tetraanion undergoes a significant geometrical reorganization.
The second example is offered by the 181-CVE monoanion [Ir14(CO)27]4-, which is related to the preceding Ir12 dianion, in that it possesses the complete (n2) trigonal-bipyramidal geometry [8].
As illustrated in Fig. 8, the closed molecular arrangement of the Ir14 monoanion is sufficiently flexible to sustain reversibly either the [Ir14(CO)27]4-/ [Ir14(CO)27]0 oxidation or the [Ir14(CO)27]4-/ [Ir14(CO)27]2- reduction [8].
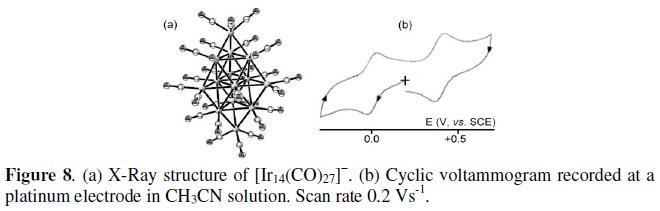
The formal electrode potentials of the electron transfer processes exhibited by the Ir12 and Ir14 clusters are reported in Table 2.
Platinum clusters
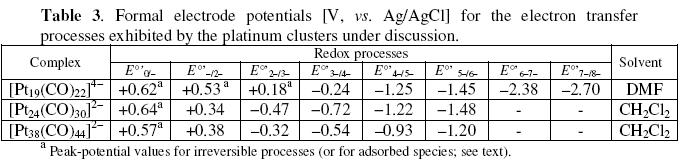
The high-nuclearity platinum clusters [Pt19(CO)22]4- , [Pt24(CO)30]2- and [Pt38(CO)44]2- exhibit an unexpected high aptitude to undergo reversible redox processes.
Let us start with the 230-CVE tetraanion [Pt19(CO)22]4-. Its molecular structure consists of a stacked sequence of three pentagonal layers of platinum atoms, sandwiching two platinum atoms, and capped at opposite sides by one platinum atom, Fig. 9a [9]. As shown in Fig. 9b, it undergoes a sequence of close-spaced pairs of reversible one-electron processes, stepwise ranging from 0 to 8- overall charge [10].
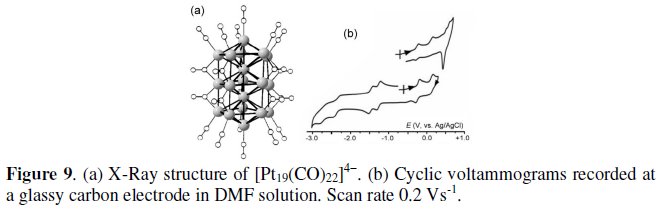
It is noted that the electrogenerated neutral species [Pt19(CO)22]0 undergoes adsorption to the electrode surface.
The pertinent redox potentials of the redox changes exhibited by the cluster are reported in Table 3, together with those of the mentioned platinum clusters discussed below.
As far as the 302-CVE dianion [Pt24(CO)30]2- is concerned, Fig. 10a shows its crystal structure, which has a cubic closest packing of Pt atoms constituted by four parallel planes of Pt atoms (4, 9, 8, 3); the nine platinum atoms layer contains an interstitial Pt atom [11].
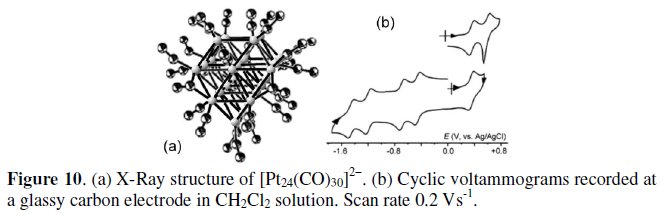
The pertinent cyclic voltammetric profile shown in Fig. 10b indicates that it also undergoes reversibly a sequence of one- electron process, namely two oxidations and four reductions, according to the stepwise passage of the overall charge from 0 to 6-; see Table 3 [10, 11].
Also in this case, the electrogenerated neutral species [Pt24(CO)30]0 is adsorbed at the electrode surface.
Finally, we discuss about the 470-CVE dianion [Pt38(CO)44]2-, whose molecular structure, which is illustrated in Fig. 11a, consists of an overall n3 truncated octahedron [12]. As shown in Fig. 11b, its redox profile is reminiscent that of [Pt24(CO)30]2-, in that it also undergoes reversibly the progressive passage of the overall charge from 0 to 6-; see Table 3 [10, 11].
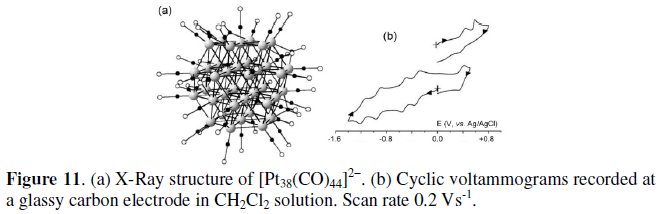
Osmium clusters
We like to discuss shortly about the dianion [Os20(CO)40]2-, the macro-tetrahedral geometry of which is shown in Fig. 12a [13]. Unpublished cyclic voltammetric data revealed a rich electrochemistry, which, based on the previously described behavior of platinum clusters, have been now tentatively reinterpreted according to the profile illustrated in Fig. 12b.
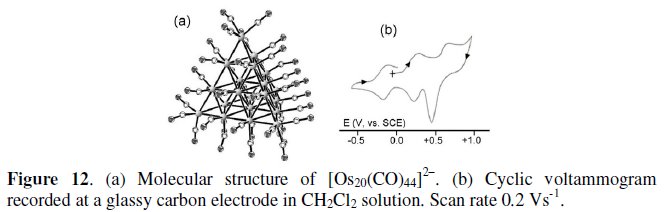
It undergoes reversibly the oxidations [Os20(CO)44]>sup>2-/[Os20(CO)44]- (E0' = +0.32 V) and [Os20(CO)44]-/[Os20(CO)44]0 (Ep = +0.74 V, accompanied by electrode adsorption of the neutral species) and the reduction [Os20(CO)44]>sup>2-/[Os20(CO)44]3- (E0' = -0.15 V).
Nickel clusters
Concerned with extended the electrochemical activity of high-nuclearity Nicluster, two Ni-carbide deserve attention: [Ni32C6(CO)36]6- and [Ni38C6(CO)42]6-.
Fig. 13 shows the truncated octahedral geometry of the 410-CVE hexa-anion [Ni32C6(CO)36]6- and its voltammetric behavior in MeCN solution [14].
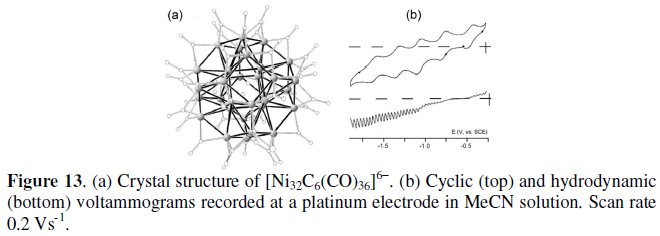
As seen, joint cyclic and hydrodynamic voltammetric techniques allow detecting that the Ni32 cluster undergoes four sequential reductions and one oxidation possessing features of chemical reversibility in the short timescale of cyclic voltammetry. Nevertheless, under the longer times of macroelectrolysis, only [Ni32C6(CO)36]7- appears perfectly stable.
The formal electrode potentials of the mentioned redox processes are summarized in Table 4, together with those of the related [Ni38C6(CO)42]6- below discussed.
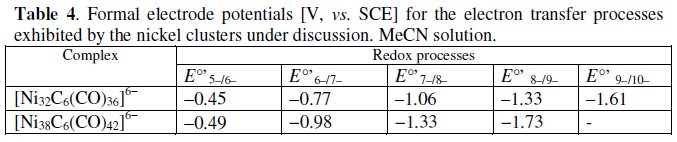
The crystal structure of the 482-CVE homologue hexa-anion [Ni38C6(CO)42]6- is available in the protonated form [HNi38C6(CO)42]5- [15]. It substantially possesses the same truncated octahedral geometry than [Ni32C6(CO)36]6- with the additional presence of six Ni(CO) units in the outer surface. Its electrochemical behavior is shown in Fig. 14 [14].
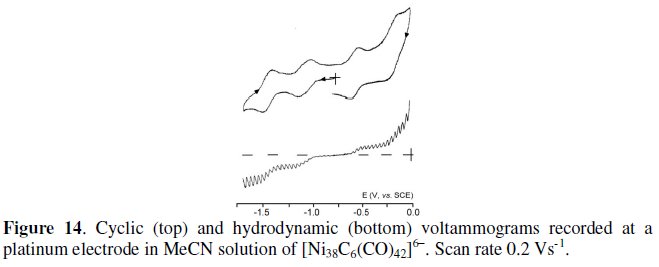
In this case, because of the cathodic shift of all the processes with respect to [Ni32C6(CO)36]6-, the accessibility to different redox steps is more limited.
Heterometallic clusters
Most uncommon chemical and physicochemical properties of heteronuclear derivatives, including metal-carbonyl clusters, arise from synergistic effects of their polar metal-metal bonds [3a].
[Fe4Au(CO)16]- vs. [Fe4Pt(CO)16]2-
The comparison of the electrochemical behavior of the isoelectronic (76-CVE) and substantially isostructural [Fe4Au(CO)16]- [16, 17] and [Fe4Pt(CO)16]2- [17, 18] illustrated in Fig. 15, is exemplificative of the unforeseen role played by the âbimetallic effectâ on the redox activity of metal carbonyl clusters.
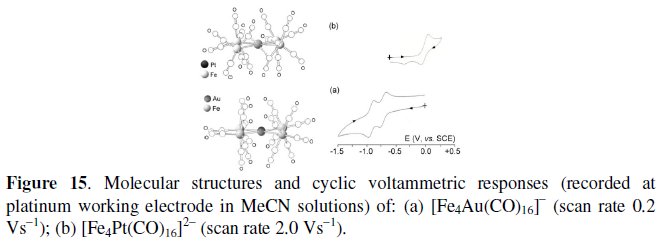
As seen, the Fe4Au monoanion undergoes reversibly the sequence -/2- (E0' = -0.73 V, vs. SCE) and 2-/3- (E0' = -0.93 V), whereas the Fe4Pt dianion only undergoes the partially chemically reversible oxidation 2-/- (E0' = +0.01 V).
[Fe3Pt3(CO)15]2-
The raft-like geometry of the 86-CVE dianion [Fe3Pt3(CO)15]2- is illustrated in Fig. 16 [17,19] together with its cyclic voltammetric response [20].
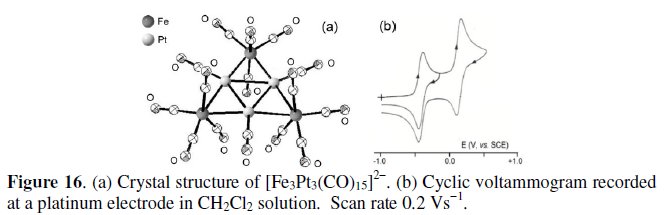
It reversibly undergoes the sequential oxidation processes [Fe3Pt3(CO)15]2-/1-/0 at potential values: E0' = +0.19 V and -0.40 V, respectively; V vs. SCE. In confirmation, the crystal structures of both the monoanion and the neutral congeners are available, [19, 21] and a few pertinent bond lengths are reported in Table 5.
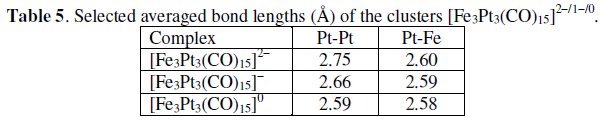
The significant shortening of the Pt-Pt distances and the substantially unchanged Pt-Fe distances indicate that the HOMO of the dianion is antibonding with respect to the three platinum atoms.
[Co10RhN2(CO)21]3- vs. [Co11N2(CO)21]3-
An example concerned with the unforeseen role of the synergistic effects of heterometallic vs. homometallic derivatives is offered by the 154-CVE species [Co10RhN2(CO)21]3- and the already examined [Co11N2(CO)21]3-.
Fig. 17 gives an overall picture of the structure and electrochemistry of the two derivatives.
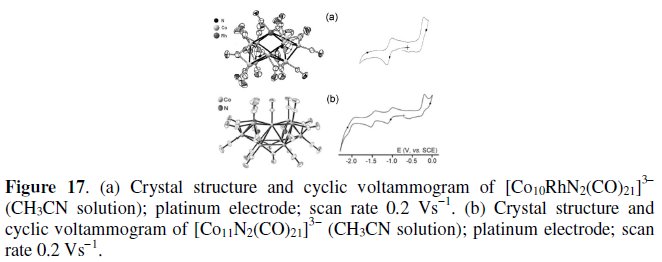
The metal frame of [Co10RhN2(CO)21]3- can be seen as composed of two capped trigonal prisms (each containing an interstitial nitrido atom) that share a triangular face [22], whereas, as already discussed in section 2.1, [Co11N2(CO)21]3- is composed by two octahedrons having a common vertex (each containing an interstitial nitrido atom) [5b].
The comparison with [Co11N2(CO)21]3- (which undergoes three one-electron reductions and a two-electron oxidation; see Table 1), shows that [Co10RhN2(CO)21]3- has a more limited electron transfer aptitude. In fact, it displays either a single one-electron reduction (E0' = -1.06 V) or an apparent two-electron oxidation (E0' = -0.24 V), both complicated by chemical reactions [22].
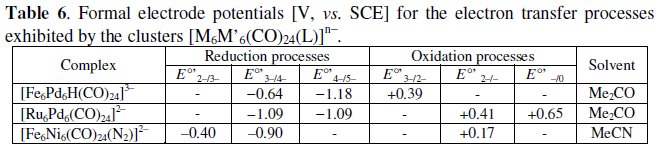
[M6M'6(CO)24(L)]n-
With the general formula [M6M'6(CO)24(L)]n-, we intend to include [Fe6Pd6(CO)24(H)]3-, [Fe6Ni6(CO)24(N2)]2- and [Ru6Pd6(CO)24]2-.
As schematized in Fig. 18, the isostructural [Fe6Pd6H(CO)24]3- (160-CVE) [18] and [Ru6Pd6(CO)24]2- (158-CVE) [23], are constituted by a trigonally distorted octahedron of Pd atoms capped by six Fe/Ru atoms.

[Pd6Ru6(CO)24]2- displays two chemically reversible one-electron oxidations, whereas [Fe6Pd6H(CO)24]3- exhibits only a single oxidation. In both cases, the chemical stability of the one-electron oxidation has been proved by controlled potential coulometry. As far as the cathodic behavior is concerned, [Fe6Pd6H(CO)24]3- exhibits two one-electron reductions, that are coupled to slow chemical complications. In turn, [Pd6Ru6(CO)24]2- apparently also gives rise to two reduction processes coupled to chemical complications, but, in reality, as deducible from the voltammogram at high scan rates shown in the inset, it gives rise to a reversible two-electron reduction affording the short lived congener [Pd6Ru6(CO)24]4-.
The formal electrode potentials of the mentioned redox changes are compiled in Table 6, together with those of the below discussed dianion [Fe6Ni6(CO)24(N2)]2-.
Finally, Fig. 19a shows the crystal structure of the 168-CVE dianion [Fe6Ni6N2(CO)24]2- (in which the metal frame forms three octahedral cavities, the two external being filled by the two interstitial nitrides), together with its cyclic voltammetric response in MeCN solution, Fig. 19b [24].
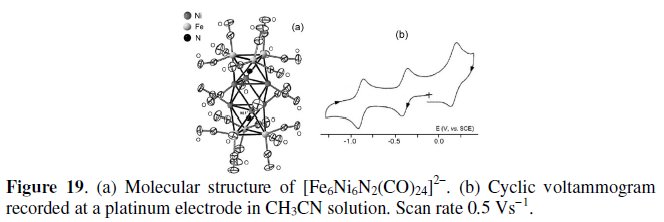
The dianion exhibits a (coulometrically measured) one-electron oxidation, which possesses features of partial chemical reversibility, as well as two stepwise (coulometrically measured) one-electron reductions, which are chemically reversible both in the cyclic voltammetric and in the macroelectrolysis timescales, Table 6.
In agreement with the chemical reversibility of the reduction processes, the crystal structures of the members of the whole sequence [Fe6Ni6N2(CO)24]2-/3-/4- have been solved. As expected on the basis of the electrochemical reversibility, the variations in metal-metal bonds are rather limited, Table 7.
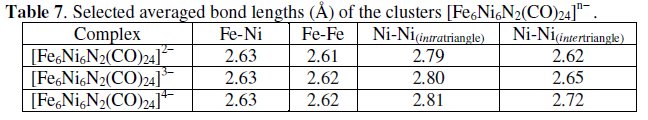
The fact that only the Ni-Ni bonds monotonically increase with the increase of the number of valence electrons, suggests that the electron additions enter the central octahedron and populate an antibonding orbital.
[Co8Pt4C2(CO)24]2-
The crystal structure of the 170-CVE dicarbide dianion [Co8Pt4C2(CO)24]2- and its cyclic voltammetric pattern are presented in Fig. 20 [25].
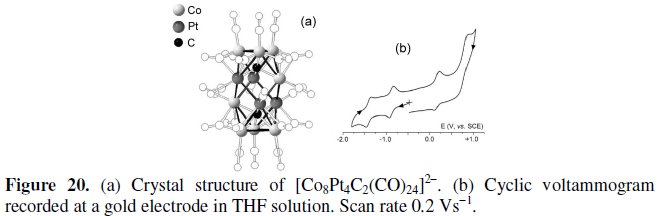
The molecular structure closely resembles those previously reported for [Co11RhN2(CO)24]2- and [Fe6Ni6N2(CO)24]n- (n = 2-4), in that, its metal cage consists on a face-sharing trioctahedron in which the two external assemblies encapsulate a carbide atom.
From the electrochemical viewpoint, [Co8Pt4C2(CO)24]2- undergoes two reduction processes possessing features of chemical reversibility (E0' = -0.91 V and E0' = -1.38 V, vs. SCE) and two oxidation processes, only the first one being chemically reversible (E0 = +0.18 V), whereas the most anodic process is coupled to chemical complications (E0' = +0.77 V).
As a matter of fact the monoanion [Co8Pt4C2(CO)24]- has been crystallographically characterized [25] and a few significant structural parameters are reported in Table 8.
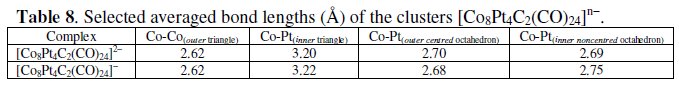
As seen, as in the case of [Fe6Ni6N2(CO)24]n-, the fragment involved in the electron transfer process is the inner octahedron.
[Ag13Fe8(CO)32]3-
The molecular structure of the 226-CVE trianion [Ag13Fe8(CO)32]3-, which is illustrated in Fig. 21, consists of a centered cuboctahedron of silver atoms with the triangular faces capped by Fe(CO)4 units [26].
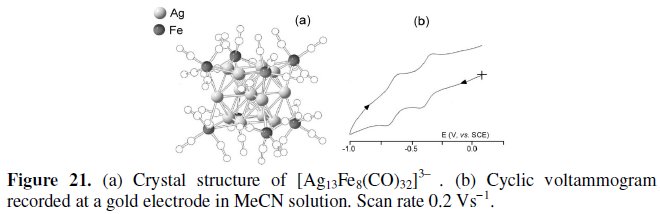
The pertinent cyclic voltammogram, Fig. 21b, shows that the trianion in MeCN solution undergoes reversibly the reduction to the corresponding [Ag13Fe8(CO)32]4- (E0' = -0.37 V, vs. SCE) and [Ag13Fe8(CO)32]5- (E0' = -0.65 V). As a consequence, such tetra-and penta-anions have been structurally characterized [27,28] and a few bond lengths for the sequence [Ag13Fe8(CO)32]3-/4-/5- are summarized in Table 9.
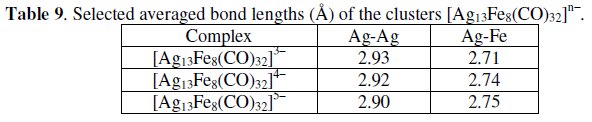
Since the distances Ag-Ag tend to decrease and the Ag-Fe tend to increase with the addition of electrons, it is evident that the LUMOs of the cluster are bonding with respect to the Ag-cuboctahedron and antibonding with respect to the Fe-capping subunits.
[Os18Pd3(C)2(CO)42]2-
The 268-CVE dicarbide dianion [Os18Pd3(C)2(CO)42]2- possesses the quite spectacular structure shown in Fig. 22a, which can be described as a triangular Pd3 metal unit sandwiched between two tricapped octahedral {Os9(μ6-C)(CO)21} subclusters [29].
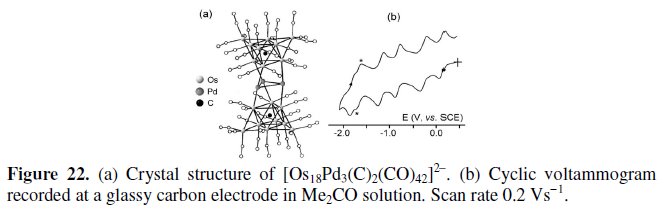
As illustrated in Fig. 22b, the dianion undergoes five reversible reductions at E0' = +0.17 V, -0.21 V, -0.77 V, -1.20 V, -1.71 V (V, vs. SCE). The asterisked reduction process at -1.63 V is attributed to the [N(PPh3)2]+ counterion.
We note that the present cluster is isostructural with the 274-CVE dianion [Os18Hg3(C)2(CO)42]2- [30], which however possesses a markedly lower redox activity [31].
[Ni38Pt6(CO)48]6-
As illustrated in Fig. 23a, the metal frame of the 542-CVE hexaanion [Ni38Pt6(CO)48]6- can be represented by an octahedron of platinum atoms encapsulated in a (ν3) macro-tetrahedron constituted by 38 nickel atoms. Such a geometrical organization is structurally proved by crystal structure of the protonated congener [Ni38Pt6(CO)48]5- [32].
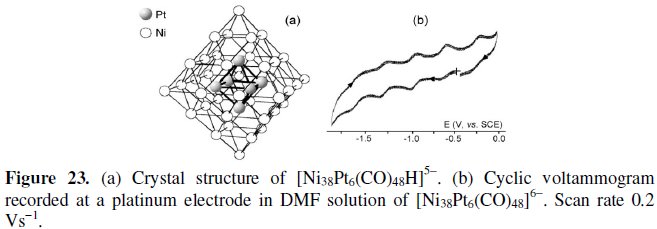
As shown in Fig. 23b, the hexaanion displays a rich redox aptitude exhibiting five one-electron redox processes [namely, one oxidation (E0' = -0.62 V, vs. SCE) and four reductions (E0' = -0.97 V, -1.29 V, -1.54 V and -1.75 V, respectively)] having features of chemical and electrochemical reversibility in the cyclic voltammetric time scale.
Conclusion
We have focused on the electrochemical behavior of a series of high-nuclearity metal-carbonyl clusters able to give rise to sequences of reversible, one-electron transfer processes in order to attest their potential use in molecular electronics. In particular, we tried to support their possible use as nanocapacitors proving that they afford consecutive redox processes separated by finite differences in potential values. Such a parameter, preluding to their semiconducting nature, is crucial to the accurate control of the flow of electrons needed to charge the device. The event has not to be neglected in that in conducting materials the capacitor charging proceeds through an uncontrolled cascade of electrons. In fact, we gave evidence that the transition from semiconductor to metallic conductor takes place for metal-carbonyl clusters having nuclearities above a likely threshold value of ca. 70 [33].
References
1.(a) C. Femoni, M.C. Iapalucci, F. Kaswalder et all, Coord. Chem. Rev. 250 (2006) 1580. [ Links ] (b) U. Simon, On the possibility of single electronics based on ligandstabilized metal clusters, in Metal clusters in chemistry. Vol. 3, P. Braunstein, L.A. Oro, P.R. Raithby (eds), Wiley-VCH Weinheim, (1999) pp 1342-1359. [ Links ] (c) G. Schmid, Y.P. Liu, M. Schumann et all, Nano Lett. 1 (2001) 405. [ Links ] (d) S. Hoeppener, L. Chi, H. Fuchs, Nano Lett. 2 (2002) 459. [ Links ] (e) M.H.V. Werts, M. Lambert, J-P. Bourgoin, M. Brust, Nano Lett. 2 (2002) 43. (f) A.C. Templeton, W.P. Wuelfing, R.W. Murray, Acc. Chem. Res. 33 (2000) 27. [ Links ] (g) L.F. Chi, M. Hartig, T. Drechsler et all, Appl. Phys. Lett. 66 (1998) S187. (h) R.L. Carroll, C.D. Gorman, Angew. Chem. Int. Ed. 41 (2002) 4378. [ Links ] (i) J. Sinzig, L.J.de Jongh, A. Cerotti et all, Phys. Rev. Lett. 81 (1998) 3211.
2.(a) D. Collini, C. Femoni, M.C. Iapalucci et all, Angew. Chem. Int. Ed. 41 (2002) 3685. [ Links ] (b) V. G. Albano, F. Demartin, M.C. Iapalucci et all, J. Chem. Soc., Dalton Trans. (1991) 739. [ Links ]
3. (a) P. Zanello, Stereochemical aspects associated with the redox behavior of heterometal carbonyl clusters, Struct. Bonding (Berlin) 79 (1992) 101. [ Links ] (b) P. Zanello, The redox propensity of high nuclearity metal clusters "NATO ASI Series", Series C: Mathematical and Physical Sciences, Kluwer Academic Publishers, Vol. 385, 1993, p. 229. [ Links ] (c) P. Zanello, Stereochemical aspects of the redox propensity of homometal carbonyl clusters, in "Stereochemistry of Organometallic and Inorganic Compounds", P. Zanello, ed. Elsevier, Amsterdam; Vol. 5, (1994) 163-408. [ Links ] (d) P. Zanello, F.F. de Biani, Bimetallic effects on the redox activity of transition-metal carbonyl clusters, in Metal Clusters in Chemistry, P. Braunstein, L.A. Oro, P.R. Raithby, Eds., Wiley-VCH, Vol. 2, 1999, 1104-1136. [ Links ] (e) G. Longoni, C. Femoni, M.C. Iapalucci, P. Zanello, Electron-sink features of homoleptic transition-metal carbonyl clusters, in Metal Clusters in Chemistry, P. Braunstein, L.A. Oro, P.R. Raithby Eds., Wiley-VCH, Vol.2, 1999, 1137-1158. (f) P. Zanello, Structure and electrochemistry of transition metal carbonyl clusters with interstitial or semi-interstitial atoms: contrast between nitrides or phosphides and carbides, in Physical Organometallic Chemistry. Vol. 3: Unusual Structures and Physical Properties in Organometallic Chemistry. M. Gielen, R. Willem, B. Wrackmeyer, eds., John Wiley, Chichester, UK., 2002, pp. 1-49. [ Links ]
4. G. Ciani, A. Sironi, S. Martinego et all, Inorg. Chem. 40 (2001) 3905. [ Links ]
5.(a) A. Fumagalli, S. Martinengo, M. Tasselli et all, Inorg. Chem. 37 (1998) 2826. [ Links ] (b) A. Fumagalli, P. Ulivieri, M. Costa et all, Inorg. Chem. 43 (2004) 2125. [ Links ]
6.(a) V.G. Albano, D. Braga, P. Chini et all, J. Chem. Soc., Dalton Trans. (1982) 645. [ Links ] (b) A. Fumagalli, M. Costa, R.D. Pergola et all, Inorg. Chim. Acta 350 (2003) 1006. [ Links ]
7. R.D. Pergola, F. Demartin, L. Garlaschelli et all, Inorg. Chem. 32 (1993) 3670. [ Links ]
8. R.D. Pergola, L. Garlaschelli, M. Manassero et all, Angew. Chem., Int. Ed. Engl. 32 (1993) 1347. [ Links ]
9. D.M. Washecheck, E.J. Wucherer, L.F. Dahl et all, J. Am. Chem. Soc. 101 (1979) 6110. [ Links ]
10. S. Fedi, P. Zanello, F. Laschi et all, J. Solid State Electrochem. 13 (2009) 1497. [ Links ]
11.(a) G.J. Lewis, J.D. Roth, R.A. Montag et all, J. Am. Chem. Soc. 112 (1990) 2831. [ Links ] (b) J.D. Roth, G.J. Lewis, L.K. Safford et all, J. Am. Chem. Soc. 114 (1992) 6159. [ Links ]
12. A. Ceriotti, N. Masciocchi, P. Macchi, G. Longoni, Angew. Chem. Int. Ed. 38 (1999) 3724. [ Links ]
13. A.J. Amoroso, L.H. Gade, B.F.G. Johnson et all, Angew. Chem, Int. Ed. Engl. 30 (1991) 107. [ Links ]
14. F. Calderoni, F. Demartin, F.F. de Biani et all, Eur. J. Inorg. Chem. (1999) 663. [ Links ]
15. A. Ceriotti, A. Fait, G. Longoni et all, J. Am. Chem. Soc. 108 (1986) 8091. [ Links ]
16. V.G. Albano, R. Aureli, M.C. Iapalucci et all, J. Chem. Soc., Chem. Commun. (1993) 1501. [ Links ]
17. C. Femoni, M.C. Iapalucci, G. Longoni et all, Chem. Eur. J. 13 (2007) 6544. [ Links ]
18. G. Longoni, M. Manassero, M. Sansoni, J. Am. Chem. Soc. 102 (1980) 3242. [ Links ]
19. G. Longoni, M. Manassero, M. Sansoni, J. Am. Chem. Soc. 102 (1980) 7973. [ Links ]
20. R.D. Pergola, L. Garlaschelli, C. Mealli et all, J. Cluster Sci. 1 (1990) 93. [ Links ]
21. R.D. Adams, I. Arafa, G. Chen, Organometallics 9 (1990) 2350. [ Links ]
22. M. Costa, R.D. Pergola, A. Fumagalli et all, Inorg. Chem. 46 (2007) 552. [ Links ]
23. E. Brivio, A. Ceriotti, R.D. Pergola et all, J. Chem. Soc., Dalton Trans. (1994) 3237. [ Links ]
24. R.D. Pergola, M. Bruschi, F.F. de Biani et all, C.R. Chimie 8 (2005) 1850. [ Links ]
25. C. Femoni, M.C. Iapalucci, G. Longoni et all, J. Am. Chem. Soc. 132 (2010) 2919. [ Links ]
26. V.G. Albano, F. Calderoni, M.C. Iapalucci et all, J. Cluster Sci. 6 (1995) 107. [ Links ]
27. V.G. Albano, L. Grossi, G. Longoni et all, J. Am. Chem. Soc. 114 (1992) 5708. [ Links ]
28. D. Collini, C. Femoni, M.C. Iapalucci, G. Longoni, C.R. Chimie 8 (2005) 1645. [ Links ]
29. K.-F. Yung, W.-T. Wong, Angew. Chem. 115 (2003) 571. [ Links ]
30. L.H. Gade, B.F.G. Johnson, J. Lewis et all, J. Chem. Soc., Chem. Commun. (1990) 110. [ Links ]
31. L.H. Gade, B.F.G. Johnson, J. Lewis et all, J. Chem. Soc., Dalton Trans., (1992) 3249. [ Links ]
32. A. Ceriotti, F. Demartin, G. Longoni et all, Angew. Chem., Int. Ed. Engl. 24 (1985) 696. [ Links ]
33. F.F. de Biani, C. Femoni, M.C. Iapalucci et all, Inorg. Chem. 38 (1999) 3721.
Acknowledgements
Piero Zanello gratefully acknowledges the financial support of the Italian MIUR (PRIN 2008).
* Corresponding author. E-mail address: zanello@unisi.it
Received 21 December 2010; accepted 3 January 2011

