










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.32 no.1 Coimbra jan. 2014
https://doi.org/10.4152/pea.201401051
Voltammetric Analysis of Montelukast Sodium in Commercial Tablet and Biological Samples Using the Hanging Mercury Drop Electrode
Ali F. Alghamdi*
Department of Chemistry, College of Science, Taibah University, P.O. Box 30002, Madinah, Saudi Arabia
Abstract
Adsorptive stripping voltammetry was used to prospect the adsorption property of montelukast sodium (MKST) on the hanging mercury drop electrode (HMDE). Through appointing the adsorptive stripping voltammetric (AdSV) process, a sensitive electroanalytical method for the quantitative analysis of MKST was accomplished. A well-developed voltammetric peak was obtained in pH 10 Britton-Robinson buffer (B-R buffer) at -1.080 V. The cyclic voltammetric studies indicated that the reduction process was irreversible and primarily controlled by adsorption phenomena. The studies of the variation of adsorptive voltammetric peak current with buffer electrolyte, pH, accumulation time (tacc), accumulation potential (Eacc), sweep rate, pulse amplitude, square wave frequency, working electrode area and convection rate have evaluated in the recognition of optimal experimental conditions for MKST analysis. The studied electroanalytical signal showed a linear response for MKST in the concentration range 5×10-8 - 1×10-6 mol L-1 (r = 0.994). A detection limit of 4×10-9 mol L-1 with relative standard deviation of 1.1 RSD% and mean recovery of 102±2.0% were obtained. The possible interferences by several compounds usually present in the pharmaceutical formulation were also evaluated. The analytical quantification of MKST drug in commercially available pharmaceutical formulation and biological samples was electrochemically studied.
Keywords: adsorptive stripping voltammetry, cyclic voltammetry, montelukast sodium, hanging mercury drop electrode, pharmaceutical formulation.
Introduction
Generally, electrochemical techniques that measure or monitor the current flow between a pair of electrodes utilize the electrolyte cell concept. This current is proportional to the concentration of the analyte in the solution. Voltammetric methods measure the current that flows as the applied potential is varied [1]. Adsorptive stripping voltammetry (AdSV) method has been well characterized as an extremely sensitive source for electroanalytical measurements since its establishment half a century ago. Such electrochemical approach with improved sensitivity and selectivity have promoted the development of numerous analytical applications of ultra-trace determinations of a variety of organic or inorganic substances. AdSV method involves a stripping step carried out by using a square wave time-potential waveform imposed on the working electrode. The principle advantages of AdSV over other stripping techniques (namely differential pulse, linear sweep and so forth) are its enhanced powers of detection, velocity of analysis and sensitivity of dissolved oxygen in the analysed samples [2-4]. Cyclic voltammetry belongs to the category of voltammetric techniques based on a linear potential sweep chronoamperometric techniques. It certainly constitutes the most useful technique for a preliminary determination of the redox properties of a given species [5].
There have been many reviews devoted to emphasize and illustrate the wide spectrum and scope of AdSV applications and potentialities in the analysis of metal ions [6-10], food analysis [11], organic analytes [12,13] and pharmaceutical drugs and biomedical compounds [14-25].
Montelukast (trade names Singulair and Montelo-10) is a leukotriene receptor antagonist (LTRA) used for the maintenance treatment of asthma and to relieve symptoms of seasonal allergies. It is used for a number of conditions such as: asthma, exercise induced bronchospasm, allergic rhinitis, and urticaria. Its discovery had a significant impact on treatment strategies for the management of asthma. It is mainly used as a complementary therapy in adults in addition to inhaled corticosteroids, if they alone don't bring the desired influence. Corticosteroids lessen inflammation but have no effect on the leukotrienes. Mountelukast sodium is described chemically as [R-(E)]-1-[[[1-[3-[2-(7-chloro2quinolinyl) ethenyl]phenyl]-3-[2-(1-hydroxy-1-methylethyl) phenyl] propyl] thio] methyl] cyclo propane acetic acid, mono-sodium salt. The chemical formula of MKST is C35H35ClNNaO3S, and its molecular weight is 608.18 [26-29]. The structural formula of this analysed pharmaceutical compound is exhibited in scheme 1.
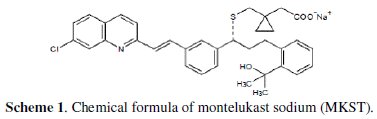
MKST has been analyzed in pharmaceutical formulations and biological samples by various analytical methods such as spectrophotometry [30,31], high performance liquid chromatography (HPLC) [32,33] and electrochemical methods such as polarography and differential pulse voltammetry [34]. However, no literature data were found on the square wave -adsorptive stripping voltammetry in general and of this drug in particular. Consequently, the development of SW-AdSV method for the analysis of MKST, and its application to determination in pharmaceutical formulations and biological fluids, is described.
Experimental
Apparatus
All electrochemical measurements were carried out with a 797 VA (Metrohm, Switzerland) connected with Dell computer and controlled by (VA computrace 2.0) control software. The stripping voltammograms were printed via a hp color laserjet CP1215 printer. A conventional three electrode system was used in the hanging mercury drop electrode (HMDE) mode. The pH values were measured with a Hanna instruments pH211 (Romania made). Oxford adjustable micropipette (Ireland) was used to measure microliter volumes of the standard solutions. The labofuge 200 instrument, Heraeus sepatech (German made) was used to centrifuge the biological fluids to be suited for stripping analysis.
Reagents
Montelukast sodium stock solution (Qassim Pharmaceutical Plant, Buraydah, Spimaco Addwaeih, Saudi Arabia) of 1×10-3 mol L-1 was prepared by dissolving the appropriate amount of MKST in distilled water in a 50 mL volumetric flask and this stock solution was stored in the dark. Britton-Robinson supporting buffer (pH ≈ 2, 0.04M in each constituent) was prepared by dissolving 2.47 g of boric acid (winlab, UK) in 500 mL distilled water containing 2.3 mL of acetic acid (BDH, UK), then adding 2.7 mL of phosphoric acid (Riedal-deHaen, Germany) and diluting to 1000 mL with distilled water. In addition, phosphate buffer [0.1 M NaH2PO4(winlab, UK) and 0.1 M H3PO4] was prepared by dissolving 12 g of NaH2PO4 and 6.78 g of H3PO4 in 1000 mL H2O. Acetate buffer (0.02 M in each constituent) was prepared by dissolving 1.68 g of sodium acetate (BDH, UK) in 500 mL H2O containing 1.12 mL of acetic acid and diluting to 1000 mL with H2O. While, carbonate buffer (0.1 M in each constituent) was prepared by dissolving 10.6 g of sodium carbonate (winlab, UK) and 8.4 g of sodium hydrogen carbonate (winlab,UK) in 1000 mL distilled water.
Procedure
The general procedure adopted for getting adsorptive stripping voltammograms was as follows: a 10 mL of B-R supporting electrolyte at desired pH was pipetted in a clean and dry electrochemical cell and the required standard solutions of MKST were added. The test solutions were purged with nitrogen for 5 minutes initially, while the solution was stirred. The accumulation potential of -0.4 V vs. Ag/AgCl was applied to a new mercury drop while the solution was stirred for 180 seconds. Following the preconcentration period, the stripping was stopped and after 10 seconds had elapsed, and cathodic scans were carried out over the range 0.0 to -1.3 V. All measurements were carried out at room temperature.
Application to tablets
The tablet solution was prepared by use five tablets weighed accurately and finely powdered and mixed. The tablets were given stock solution of MKST (1×10-3 mol L-1) in 50 mL of MeOH by use volumetric flask. The content of the flask was left in the room temperature and the dark for 48 h to provide dissolution and then completed to forthwith determine MKST compound by use of the standard additions method.
Application to biological fluids
Regarding human urine and plasma preparations, 1.0 mL of 5% ZnSO4.7 H2O solution, 0.1 mL of NaOH and 1.0 mL of ethanol were added to 0.5 mL of human urine and plasma samples and 3×10-7 mol L-1 MKST drug [35]. This solution was centrifuged for 15 min at 4000 rpm to separate out the insoluble excipients. Thereafter, the solution was taken and diluted B-R buffer at pH 10 to achieve the desired concentration. Then the procedure was completed by use standard additions method.
Results and discussion
The electroanalytical properties of MKST
The preliminary voltammetric study of MKST pharmaceutical molecule indicates that it has cathodic reduction response onto the HMDE electrode by using differential pulse polarographic (DPP) approach and using an applied potential in the negative direction. 1×10-5 mol L-1 of MKST in pH 10 Britton-Robinson buffer was given a reduction polarographic peak, as can be seen from Fig. 1.
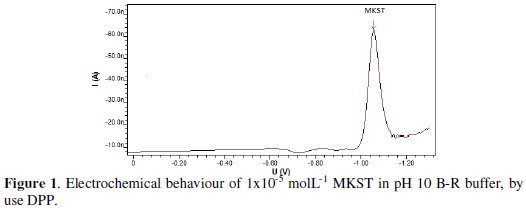
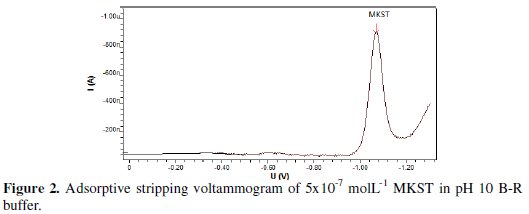
Fig. 2 exhibited a stripping reduction voltammogram for 5×10-7 mol L-1 MKST in pH 10 B-R buffer by using square wave voltammetric technique.
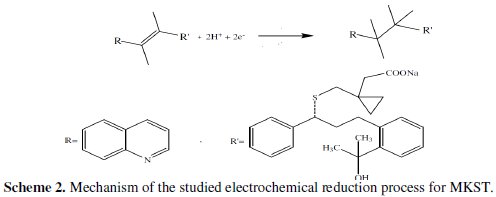
However, the addition of 5×10-7 mol L-1 MKST to the test solution provided a well-defined cathodic peak at -1080 mV (versus Ag/AgCl reference electrode). The observed AdSV peak is most probably due to the cathodic reduction of the double bond (C=C) to single bond (C-C) in the sub-strain of adsorbed drug and the electrochemical mechanism of this reduction process for MKST is illustrated in scheme 2.
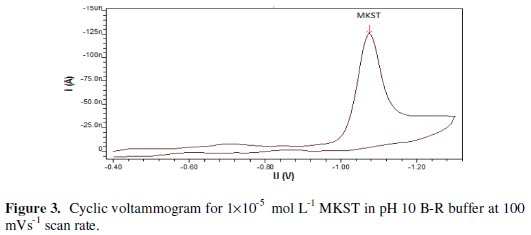
Obviously, this proposed electrochemical reduction mechanism suggested an irreversible reductive process for the adsorbed drug, an assumption which was confirmed by cyclic voltammetric measurement of 1×10-5 mol L-1 MKST drug in pH 10 B-R buffer at 100 mVs-1 scan rate. As can be noticed from Fig. 3, which exhibits the cyclic voltammogram of MKST, the absence of the anodic peak at the reverse scan confirmed the irreversible nature of the evaluated reduction process.
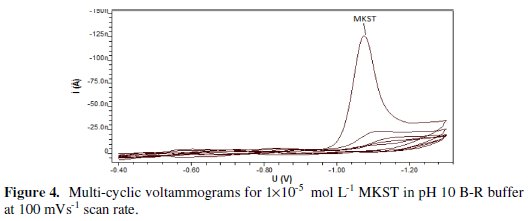
Furthermore, when repetitive cyclic voltammetric measurements for MKST drug were carried out as shown in Fig. 4, a well developed CV peak was observed at the first cathodic scan; however, succeeding cathodic scans exhibit a gradual decrease in the voltammetric peak intensity that seemed to indicate the adsorptive characteristic of this compound at the surface of the employed working electrode.
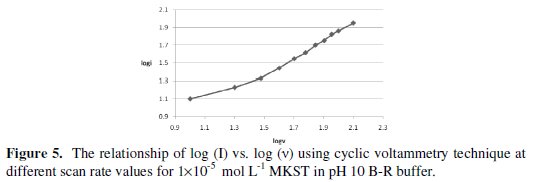
Anyway, the interfacial accumulation of this drug onto the HMDE surface can be used as an effective accumulation step in order to enhance the electroanalytical determination of MKST molecules. On the other hand, the linear enhancements between the electroanalytical signals and different values of scan rate were studied using cyclic voltammetry technique over the range 10-120 mV/s. So, a linear relationship of log(I) vs. log(v) was illustrated, as shown in Fig. 5, which yielded a slope of 0.9 that is close to 1.00 value indicating that electrochemical reduction of MKST at the working electrode (HMDE) is an adsorption-controlled process [36].
In addition, a shift to more negative potential was observed upon increasing the scan rate, which confirms the irreversibility of the electroanalytical reduction of MKST.
Optimisation of experimental parameters
Effect of supporting buffer constituents and pH
Since the adsorptive phenomenon of MKST on the working electrode (HMDE) was utilized as a suitable collection step prior to its electrochemical analysis, it was rational to characterize various variables and experimental conditions affecting the engaged adsorption procedure. Actually, the sensitivity of the adsorptive stripping process for a particular analyte is usually significantly influenced by the composition of the electrolyte buffer and pH values. Consequently, many electrolyte buffers such as: Britton-Robinson (B-R), acitae, carbonate and phosphate buffers at different pH values were evaluated after 60 sec tacc at 0.0 V Eacc. Amongst these supporting electrolytes the best electroanalytical signal in terms of AdSV peak current intensity and shape was obtained with B-R buffer, which was selected as optimal for further works.
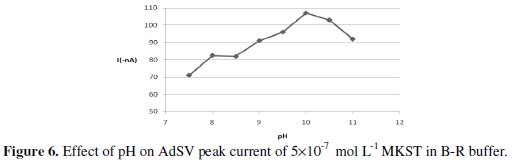
Generally, the AdSV signal was mainly pH dependent since the monitored voltammetric signal was only observed at alkaline state. When the voltammetric peak signal was measured as a function of pH over the range 7.5-11, the peak current increased gradually at first and enhanced sharply beyond pH 8.5, then it reached its maximum value at pH 10, which was adopted as the optimum pH value for subsequent investigations. The influence of pH factor on the AdSV signal is illustrated in Fig. 6.
On the other hand, it is observed that the peak potential was shifted gradually to more negative values from -670 mV to -785 mV upon increasing pH over the range 7.5-11, (see Fig. 7).
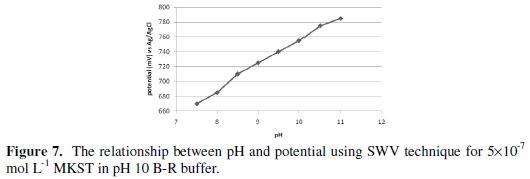
From the plot of Ep vs. pH, slope value of 33 mV/pH was recorded, that is close to the expected theoretical value of 30 mV/pH. Interestingly, this result indicates that Ep is pH dependent as expected for the electroanalytical reduction process; yield, the proposed electrochemical mechanism consumes two protons [37] for the voltammetric determination of MKST.
Effect of accumulation factors
The interfacial accumulation of MKST onto the HMDE surface depends on some factors, which added importance investigations in order to obtain high sensitive determinations for MKST drug. Therefore, the effect of accumulation time on the efficiency of the collection of 5×10-7 mol L-1 MKST onto the working electrode was evaluated by rising the accumulation time over the range 0-220 sec. The resulting peak current-accumulation time (ip-tacc) profile is exhibited in Fig. 8 and as can be seen from this plot, an enhancement in the voltammetric peak current was observed over the range 0-180 sec.
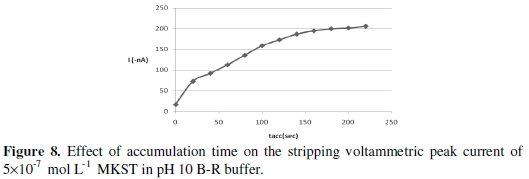
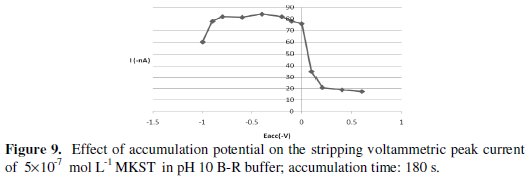
After this range, the peak intensity didn't change almost because of probably saturation of the mercury electrode. Hence, 180 sec accumulation time was selected for all additional experiments. On the other hand, variation of the accumulation potentials over the range from +0.6 V to -1.0 V at 180 sec accumulation time, as shown in Fig. 9, revealed that a pre- concentration potential of -0.4 V was the ideal choice for optimal sensitivity.
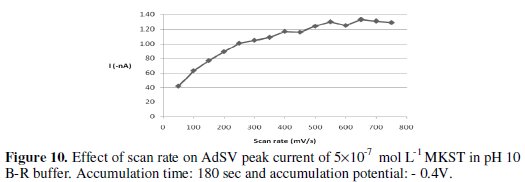
Effect of potential sweep parameters
The observed stripping voltammetric signal can be further maximized by adjusting the way the applied potential was monitored. The relationship between the peak intensity and scan rate was found to be directly proportional over 50 750 mV s-1 scan rate. However, when scan rate faster than 650 mV s-1 were used, the current decreased slightly. The effect of scan rate on the observed voltammetric signal is illustrated in Fig. 10, which indicates that scan rate value of 650 mV s-1 would be adequate optimum for succeeding investigations.
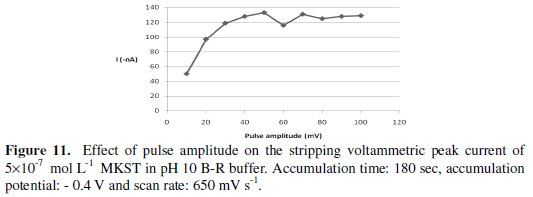
In addition, the impact of varying the excitation wave pulse amplitude on the voltammetric current intensity was evaluated. The effect of pulse amplitude was studied over the range 10-100 mV (see Fig. 11) and it was concluded that in order to assure maximum peak current, 50 mV pulse amplitude is the ideal choice for operational parameters.
In addition, varying the value of square wave frequency also plays an important role for the measured signal of AdSV approach.
Varying this parameter over the range 10-150 Hz resulted in a substantial enhancement of the voltammetric peak current, particularly at range 10-130 Hz, as can be seen from Fig. 12.
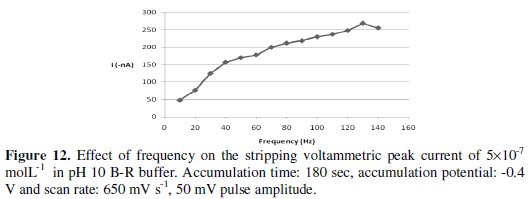
Accordingly, for future work 130 Hz SW frequency value was adopted.
Effect of other instrumental variables
The influence of other operating parameters such as the size of the adsorption area (HMDE) and convection rate on the efficiency of the adsorption accumulation of MKST was also checked. As expected, a linear enhancement relationship for the electrochemical peak intensity was observed when the surface area of HMDE was increased over the range 0.15-0.6 mm2 drop size area. Besides, the AdSV peak current can be maximized further by increasing the stirring rate of the rotating rod over the range 0-3000 rpm. Hence, for optimal sensitivity, 0.6 mm2 and 3000 rpm were selected.
In conclusion, for electroanalytical purposes, the optimised experimental conditions for AdSV measurements of MKST drug were accumulating for 180 sec at -0.4 V accumulation potential with stirring rate of 3000 rpm. These voltammetric measurements were carried out in Britton-Robinson buffer at pH 10. The applied potential was scanned at 650 mV s-1 with 130 Hz SW frequency rate and 50 mV pulse amplitude.
Analytical performance
Calibration graph and detection limit
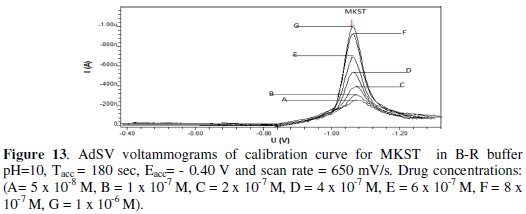
Once the optimal chemical conditions and instrumental parameters for the AdSV determination of MKST were decided, several analytical properties of the proposed were evaluated. Under the optimised conditions, a linear correlation between AdSV peak intensity and the drug concentration was obtained over the range 5×10-8 - 1×10-6 mol L-1, (see Fig. 13).
The calibration equation was calculated by least-squares method and it has the form:
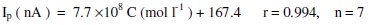
where Ip is the stripping voltammetric peak current in nano-amperes, C is MKST concentration, r is the correlation coefficient and n is number of measurements. The effective preconcentration step during the adsorption process of the analyzed drug allows a very low detectability. The detection limit estimated based on the signal-to-noise ratio (S/N = 3) was 4×10-9 mol L-1. This obtained sensitivity was significantly preferable than those reported for other analytical techniques currently used for determination of MKST drug, such as differential pulse polarography (DPP) with 3.41 ×0-7 M [34] detection limit.
Precision, accuracy and stability
The reproducibility of the developed procedure was evaluated from eight repeated measurements of 5×10-7 mol L-1 MKST. The precision of the method in terms of the relative standard deviation (RSD%) was 1.1%. The accuracy of the electrochemical method was checked by calculating the recovery of known amount (3×10-7 mol L-1) MKST drug spiked in buffer solution and analysed by the optimised procedure.
The results of five measurements obtained by the standard addition method have a recovery mean of 102% with standard deviation of ± 2.0%. When the AdSV signal of 5×10-7 mol L-1 MKST solution was monitored every ten minutes, it was found to nearly stable for a period of 2.0 hours at least. ×10-7 mol L-1
Interferences
The competitive co-adsorption interference was evaluated in the presence of various substances usually occurring in the pharmaceutical formulation as tablet ingredients or additives. For these investigations, the interfering species (lactose, sucrose, glucose and starch) were added in different concentrations (twice, 5 fold and 50 fold) higher than the MKST concentration (5×10-7 mol L-1 MKST). The addition of 2-fold, 5-fold and 50-fold of lactose, yielded negative interferences by 9.85%, 15.3% and 18.45%, respectively. Also, the addition of 2-fold, 5-fold and 50-fold of glucose, yielded negative interferences by 11.6%, 17.8% and 27.2%, respectively. Furthermore, the addition of twice, 5-fold and 50-fold of starch, yielded negative interferences by 14.7%, 20.8% and 23.8%, respectively. In contrast, the addition of 50-fold of sucrose higher than the quantity of the assayed drug caused the AdSV peak current to decrease by about 7.42% of its original signal.
This inhibition response is possibly due to the competitive co-adsorption of these interfering substances (particularly at higher concentration levels) on the adsorption sites of HMDE.
Analytical applications
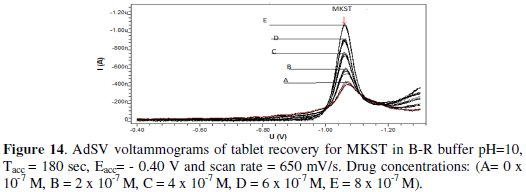
To assess the reliability of the proposed electrochemical procedure described above, it was applied for resolving the determination of MKST in pharmaceutical preparation. The drug content of the commercially available Singular tablet (Merck Sharp & Dohme Limited (MSD), Northumberland, UK: contain 10 mg of MKST) was determined via the optimized AdSV procedure directly after the required dissolving and filtration steps and the dissolved sample was diluted to the required concentration level. The electrochemical measurements were done by the standard addition method in order to minimize matrix effects. Five aliquots of this sample were analyzed by the proposed AdSV method (see recovery in Fig. 14).
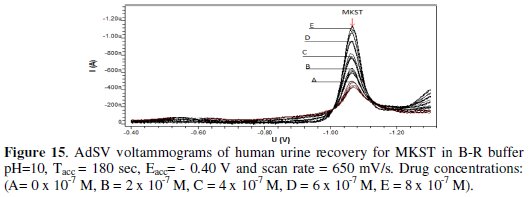
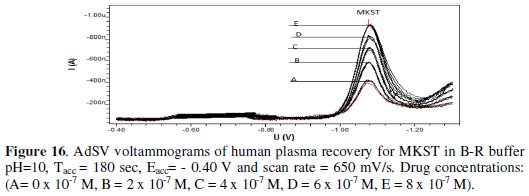
MKST was also determined in urine and plasma via recoveries as shown in Fig. 15 and 16, respectively.
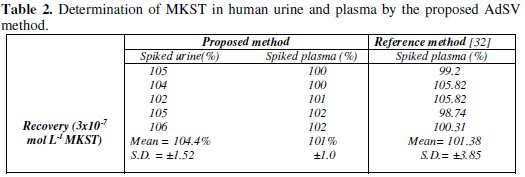
The obtained results by using the developed AdSV method with regard to determination of MKST in commercial tablet, urine and plasma are given at Tables 1 and 2.

References
1. Kenkel J. Analytical Chemistry for Technicians. 3rd . USA:CRC Lewis Publishers;2003. [ Links ]
2. Wang J. Analytical Electrochemistry. New York:VCH Publishers Inc; 1994. [ Links ]
3. Osteryoung J, O'Dea J J. In: Electroanalytical Chemistry. Vol. 14 (Bard AJ. ed.). New York:Marcel Dekker;1986. [ Links ]
4. Economou A, Filden P R. Anal Chim Acta. 1993;273.27. [ Links ]
5. Zanello P. Inorganic Electrochemistry. Theory, Practice and Application. UK:The Royal Society of Chemistry;2003. [ Links ]
6. Zaitsev P M, Salikhdzhanova R M F, Zaitsev N K. Ind Lab Diag Mater. 1999;65:1. [ Links ]
7. Abu Zuhri A Z, Voelter W. Fresenius J Anal Chem. 1998;360:1. [ Links ]
8. Achterberg E P, Braungardt C. Anal Chim Acta. 1999;400:381. [ Links ]
9. Bento F R, Grassi M T, Sales A, et al. Int J Electrochem Sci. 2008;3:1523. [ Links ]
10. Alghamdi A H. J Saudi Chem Soc. 2010;14:1. [ Links ]
11. Alghamdi A H. Arabian J. Chem. 2010;3:1. [ Links ]
12. Brainina K H, Malakhova N A, Stojko N Y. Fresenius J Anal Chem. 2000;368:307. [ Links ]
13. Manisankar P, Sundari P A, Sasikumar R. Int J Env Anal Chem. 2009;89:245. [ Links ]
14. Alghamdi A H. J. Saudi Chem Soc. 2002;6:185. [ Links ]
15. Vire J C, Kauffmann J M, Patriarche G J. J Pharma Biomed Anal. 1989;7:1323. [ Links ]
16. Alghamdi A F, Alghamdi A H, Al-Omar M A. Anal Lett. 2008;41:1. [ Links ]
17. Alghamdi A F, Alghamdi A H, Al-Omar M A. Egypt J Anal Chem. 2008;17:98. [ Links ]
18. Alghamdi A F, Alghamdi A H, Al-Omar M A. Saudi Pharm J. 2008;16:231. [ Links ]
19. Alghamdi A F, Alghamdi A H, Al-Omar M A. J Saudi Chem Soc. 2008;12:1. [ Links ]
20. Alghamdi A F, Alghamdi A H, AlOmar M A. Arabian J Chem. 2008;1:37. [ Links ]
21. Alghamdi A F, Alghamdi A H, Al Omar M A. Port Electrochim Acta. 2009;27:645. [ Links ]
22. Alghamdi J Saudi Chem Soc. 2009;13:235.
23. Alghamdi A F. Am J Anal Chem. 2011 ;2:174. [ Links ]
24. Alghamdi A F, Hefnawy M M. Arabian J Chem. 2012;5:383. [ Links ]
25. Alghamdi A F, Hefnawy M M, Al-Majed A A, et al. Chem Central J. 2012;6:15. [ Links ]
26. Budhavari S. (Ed). In: The Merck Index. New Jersey:Merck & Co Inc.;1996. P. 31-315.
27. Budhavari S. (Ed). In: The Merck Index. New Jersey:Merck & Co Inc.;1999. P. 6337. [ Links ]
28. Prafit K. Ed. In: Martindale, The complete drug reference. 33rd Ed. London:The Pharmaceutical Press;1999. P.756 [ Links ]
29. Morrow J D, Roberts L J. In: Hardman J. G., Limbrid L. E. (Eds). The Pharmacological Basis of Therapeutics. 10th Ed. New York:McGraw Hill;2001. P.677.
30. Patel D J, Patel S A, Patel S K. Int J Pharm Biomed Resear. 2010;1:71. [ Links ]
31. Arayne M S, Sultana N, Hussain F. J Anal. Chem. 2009;64:690. [ Links ]
32. Patel S, Pore Y V, Kuchecker B S, et al. Indian J Pharm Sci. 2009;71:58. [ Links ]
33. Radhakrishna T, Narasaraju A, Ramakrishna M, et al. J Pharm Biomed Anal. 2003;31:359-368. [ Links ]
34. Alsarra I, Al-Omar M, Gadkariem E A, et al. II Farmaco. 2005;60:563. [ Links ]
35. Stubauer G, Obendorf D. Analyst, 1996;121:351. [ Links ]
36. E Laviron. J Electroanal Chem. 1981;122:37.
37. Wang J. Stripping Analysis, Principles, Instrumentation and Applications. Deerfield Beach, Florida:VCH Publishers, Inc.;1985. [ Links ]
Acknowledgement
The author would like to thank Mr. Awad alqarni and Ahmad alribdy at Qassim Pharmaceutical Plant Buraydah, Spimaco Addwaeih, Saudi Arabia for their assistances and supplying us by standard drugs.
*Corresponding author. E-mail address: alifh2006@hotmail.com
Received 23 January 2014; accepted 25 February 2014

