










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Similars in
SciELO
Similars in
SciELO
Share

 Permalink
PermalinkPortugaliae Electrochimica Acta
Print version ISSN 0872-1904
Port. Electrochim. Acta vol.33 no.3 Coimbra May 2015
https://doi.org/10.4152/pea.201503183
Electrochemical Investigation of Divalent State of Praseodymium and Samarium in Non-aqueous Medium
Jignasu P. Mehta* , Kahan I. Pandya and Dinesh R. Godhani
Analytical Chemistry Division, Department of Chemistry, (UGC NON-SAP & DST-FIST sponsored Department) Mahatma Gandhi Campus, Maharaja Krishankumarsinhji Bhavnagar University, Bhavnagar-364002, India
Abstract
The aim of present study is to establish the not known divalent state of praseodymium and samarium in non-aqueous medium at 298.15 K and atmospheric pressure. Potentiostatic and galvanostatic methods were used to characterize the divalent states of both lanthanides. Under the specified experimental conditions, samarium and praseodymium showed two reduction steps at glassy carbon electrode in non-aqueous medium. Cathodic and corresponding anodic peak potential and peak currents were calculated for samarium and praseodymium ions at different scan rates. The results also suggest that the scan rate has great influence on the behaviour of both lanthanides. Diffusion coefficient (D × cm2 s-1) and heterogeneous forward rate constant (kofh × cm s-1) have been evaluated. Transition time (τ) has also been evaluated for Pr(III)/Pr(II), Pr(II)/Pr(0) coupled systems, suggesting that the system is approaching from irreversible with increasing the scan rates. The effects of changing the scan rate and donor number on the electrochemical behavior of both lanthanides have been examined at 298.15 K and atmospheric pressure.
Keywords: Praseodymium, samarium, non-aqueous medium, potentiostatic method, galvanostatic method, Sand equation.
Introduction
Recently, Ln(III) has been found to be used for various purposes in the fluorescence, luminescence, electrochemical and electroluminescence taking into account its advantages over other metal ions. However, Eu(II) is a highly reducing species [1]. There is also evidence that divalent state of europium might be useful as an indirect probe for biochemical reaction of Ca(II) ion, the most abundant metal ion in our body system but unfortunately spectroscopically silent [2]. Eu(II) was also investigated using UV-visible spectroscopy during its electro-reduction process in acidic medium [3]. One of these Eu(II) complexes is more stable than Fe(II) in hemoglobin and appears to be the most oxidative- stable aqueous Eu(II) species known. Eu(II) is expected to enable the use of the unique magnetic and optical properties of this ion in vivo. [4]. Recent advances in the coordination chemistry of Eu(II) are reviewed, the coordination chemistry of Eu(II) has led to an upsurge in the utilization of Eu(II)-containing complexes in synthetic chemistry, materials science, and medicine [5].
Recently, electrochemical studies of samarium were also reported showing that Sm(III)/Sm(II) coupled system is useful for the characterization of the matrix at high temperature [6]. Sm(II) iodide (SmI2) has experienced increasing attention since its introduction into organic chemistry by Kagan [7] some 30 years ago. Being one of the most versatile reagents for the formation of radicals utilized by chemists today, SmI2 holds a key position in the organic synthesis of fine chemicals. It has found applications in most radical transformations, including ketylâalkene couplings and cyclisations, pinacol couplings, fragmentation reactions, deoxygenations, dehalogenations, desulfonations and many others. Sm(II) iodide (SmI2) is one of the most important reducing agents in organic synthesis. It is widely used as a reduction agent and for the carbon-carbon bond forming reaction. Also, a variety of fragmentation reaction with Sm(II) iodide is known where mostly carbon heteroatom bonds are cleaved. In most cases, reactions promoted by samarium are usually carried out in THF, and metallic samarium has to be activated or pretreated by various methods to ensure smooth reactions. The most utilized and most studied ligand used to increase the reducing ability of SmI2 is hexamethylphosphoramide (HMPA). While HMPA remains as a useful additive in SmI2 mediated reactions, its carcinogenic potential outweighs its utility as a co-solvent [8-14]. Many studies have attempted to assess the significance of the use of the β-particle emitter praseodymium-142 in cancer treatment [15]. A 142Pr CTRI was proposed for brachy-therapy of prostate cancer [16]. The magnetic and optical properties of the divalent state of europium make this ion extremely attractive for use in materials [17], while samarium catalytic activity for CO2 reduction is reported [18]. Luminescence [19], magnetic [20], and diagnostic-medical applications of europium and other lanthanides are also reported. The divalent lanthanides have their own importance in the biochemistry to mimic the interaction of diamagnetic Ca(II) ion with multi-donor bio-molecules in human metabolism [21-26]. In the last 20 years, many other stable, yet very reactive, uncommon low-valent compounds have been found, including zero-valent organometallic complexes, scandium monovalent and divalent complexes, lanthanum, cerium, neodymium, dysprosium and thulium divalent complexes [27]. This is in continuation to our previous effort to characterize the divalent state of various Ln(III) ions in non- aqueous medium. The present study deals with the characterization of divalent state of praseodymium and samarium in non-aqueous medium with help of potentiostatic and galvanostatic techniques.
Experimental
Chemicals
Salts of praseodymium and samarium were purchased from Alfa-Aesar, USA with purity of 99.95% and solutions were made without purification of slats. The stock solution of each lanthanide (0.1 mol/L), tetra ethyl ammonium per chlorate (TEAP, 0.1 mol/L) were prepared in the D.W. (distilled water), acetonitrile and DMF. Further stock solutions of trivalent lanthanide (0.1 mol/L) were standardized by using standard EDTA method with a pinch of hexamine powder (G.R. grade) to maintain pH 6.0 and xylenol orange as an indicator. The final value of stock solutions of each lanthanide [Pr(III), and Sm(III)] was determined with σ = ± 0.1 (n=6) using statistical aids. The solvents used for the present study were of HPLC grade to avoid/minimize the effect of impurities associated with the solvent. The chemicals used for the present study were of analytical grade and procured from E. Merck Ltd, Spectrochem Ltd, and BDH Ltd.
Instrumental conditions
Electrochemical workstation model number 660-B with Chi software from CH Instrument, USA, was used to perform potentiostatic and galvanostatic behaviour of both Ln(III) ions. It contains a three electrodes system. We used a Saturated Calomel Electrode (SCE) as reference, a Glassy Carbon electrode (GC) as working, and a platinum electrode as counter electrode.
The saturated calomel electrode and the glassy carbon electrode have been directly purchased from CH Instrument, USA, and, according to available literature, they manufactured both electrodes as per standard protocols. The electrode surface of the glassy carbon working electrode is renewed by using a special polishing kit provided by the CH Instrument, USA, after each set of experiment, to avoid contamination at the electrode surface. Moreover, the electrode surface is renewed with 0.05-micron alumina after each individual experiment.
Methodology of the potentiostatic method
An aliquot of 1 mM of both Ln(III) ions was transferred into an electrochemical cell so that the final concentration of both ions was reached to 0.1 mM in the electrochemical cell. A suitable solvent medium was created and the resultant solution was deaerated for at least 30 minutes with dry N2 gas. The zero grade N2 gas, purchased from IOLAR & Co., was further purified by passing successively through 15% pyrogallol in 40% KOH, H2SO4 and anhydrous silica gel.
Cyclic-voltammetry was selected as a tool amongst available potentiostatic methods and cyclic voltammograms of these solutions were recorded at the glassy carbon electrode of the different potential range. The above experiments were conducted and standard methods were adopted to measure the potential and current data such as Epc, Epa, Ep/2, ipc and ipa.
To calculate the diffusion coefficient for reversible systems the following equation is used.
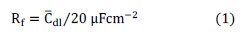
To calculate the diffusion coefficient for irreversible systems the following equation is used.
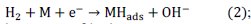
To calculate the heterogeneous forward rate constant for irreversible systems the following equation is used.
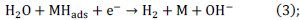
Methodology of the galvanostatic method
An aliquot of 1 mM of Sm(III) and 1 mM Pr(III) and their solutions were transferred into an electrochemical cell so that the final concentration of both ions was reached to 0.25 mM in the electrochemical cell. A suitable solvent medium was created and the resultant solution was deaerated for at least 30 minutes with dry N2 gas. The zero grade N2 gas, purchased from IOLAR & Co. was further purified by passing successively through 15% pyrogallol in 40% KOH, H2SO4 and anhydrous silica gel.
Chronopotentiometry was selected as a galvanostatic tool and chronopotentiograms were recorded at the glassy carbon electrode of the different current density range, and the optimum current density was selected to get two distinct transition times for two separate states of Sm(III) and Pr(III) in non-aqueous medium.
The Sand's equation is used for calculation of chronopotentiometric parameters:
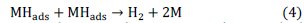
Results and discussion
The cyclic voltammetry was used as potentiostatic method and its response for all studied systems was categorized in three subclasses viz. effect of slow scan rate, effect of moderate scan rate and effect of rapid scan rate. This is because the scan rate (sweep rate) plays a very vital role in investigating the behavior of different systems from reversible to irreversible reaction at the electrode surface.
Cyclic-voltammetric study of Sm(III) in non-aqueous medium
The cyclic voltammetric responses at scan rate from 0.004 V/s to 0.02 V/s were recorded. The Sm(III)/Sm(II) coupled system in 100% ACN was investigated in the applied potential range of -0.4 Volts to -2.3 Volts with 0.1 M TEAP (μ=0.1) as supporting electrolyte. The data of Samarium were obtained to get baseline idea for divalent state of praseodymium.
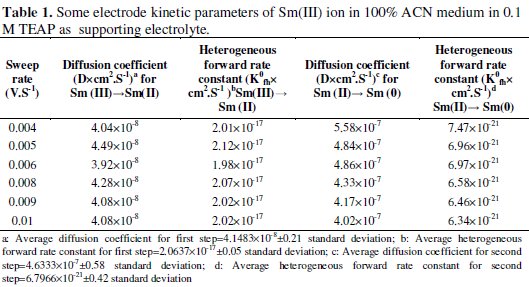
Sm(III)/Sm(II) coupled system in non-aqueous medium showed a straight line with a least square regression line passing through the origin (y = 9.118 X - 0.012; R2=0.934) suggesting that one electron transfer reaction was diffusion controlled at the electrode surface. Similarly, Sm(II)/Sm(0) coupled system in non-aqueous medium showed a straight line with a least square regression line passing through the origin (y = 3.621 X + 2.740; R2=0.958) suggesting that two electrons transfer reaction was diffusion controlled. The electrode kinetic parameters for the said system are summarized in Table 1.
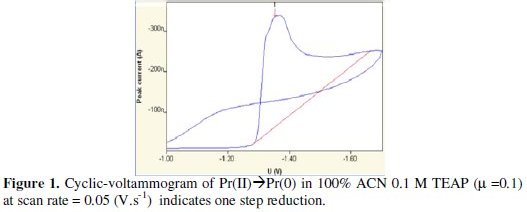
The Pr(III)/Pr(II) coupled system in 100% ACN was investigated in the applied potential range of -0.4 to -1.0 Volts with 0.1M TEAP (μ=0.1) as supporting electrolyte.
It is believed that Pr(III) generally undergoes one step reaction at electrode surface, but our results in 100% non-aqueous medium recommend that under specified experimental conditions, Pr(III) can also have two steps reduction at the electrode surface. The first step was less known to chemists; it involves one electron transfer from the bulk of the solution to the electrode surface for reduction of Pr(III) ion to Pr(II) ion, that is divalent state of praseodymium. The cathodic peak potential was varied from -0.652 V to -0.687 V with reference to SCE and the corresponding cathodic peak current was varied from 0.0108 μA to 0.0130 μA. The anodic peak was observed at -0.486 V to -0.496 V, while the corresponding anodic peak current was varied between 0.000809 μA to 0.00246 μA. The second step was observed with two electrons transfer at the electrode surface to produce the elemental praseodymium. The corresponding peak potential was varied from -1.36 V to -1.38 V with reference to calomel electrode, whereas the cathodic peak current was found to vary from 0.266 μA to 0.492 μA. The corresponding anodic plateau was not well-defined; this can be attributed to the solvent shielding around the electrode surface, which leads to slow the rate of reaction at the electrode surface to generate Pr(II) ion to Pr(III) ion. Transfer of two electrons from the divalent praseodymium to obtain elemental praseodymium was evident from the respective values of cathodic peak current, which were found almost double or more than double than those obtained for the first step. The corresponding second step is depicted in Fig. 1.
Electrode kinetic parameters were evaluated by using the modified Rendles-Sevcik, and modified Nernst equation and the results are summarized in Table 2.
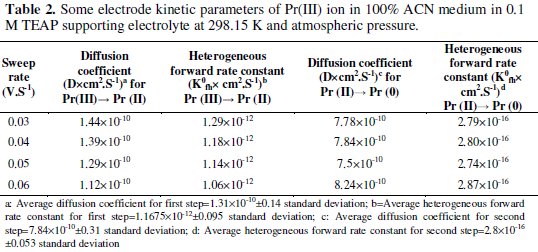
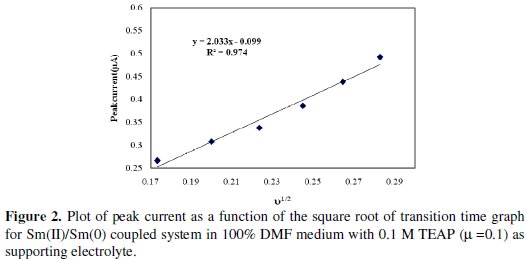
The data suggest that the rate of reaction at the electrode surface is governed by many factors such as diffusion process of Pr(III) ion from bulk of the solution to the electrode surface, possible contamination at the electrode surface, time allowed to redox coupled reaction, etc. Pr(III)/Pr(II) coupled system in non- aqueous medium showed a straight line curve with least square regression linear in nature (y = 0.021 X + 0.006; R2 = 0.949), suggesting that the one electron transfer reaction was diffusion controlled. Similarly, Pr(II)/Pr(0) coupled system in non-aqueous medium clearly indicates a straight line curve with least square regression (y = 2.033 X - 0.099; R2 = 0.974) suggesting that the two electrons transfer reaction was diffusion controlled and not governed by any other phenomenon.
Chronopotentiometric study
Chronopotentiometric technique was used as galvanostatic technique and was used to characterize the divalent state of both Sm(III) and Pr(III) ions in 100% non-aqueous medium to support the above results of cyclic-voltammetric technique. Both Sm(III) and Pr(III) were investigated by using chronopotentiometry to identify the transition time (τ) for one and two electron transfer, respectively.
Chronopotentiometry of Sm(III) ion
Under the specified experimental conditions Sm(III) was investigated for cathodic process in 0.1 M TEAP in 100% DMF medium. Chronopotentiogram of Sm(III) in the cathodic process showed a single plateau with transition time (τ) 2.55 s at 40 μA current density. Similarly, chronopotentiograms of Sm(II) to Sm(0) were recorded and are shown in the Fig. 2.
Chronopotentiometric data for samarium are summarized in Table 3.
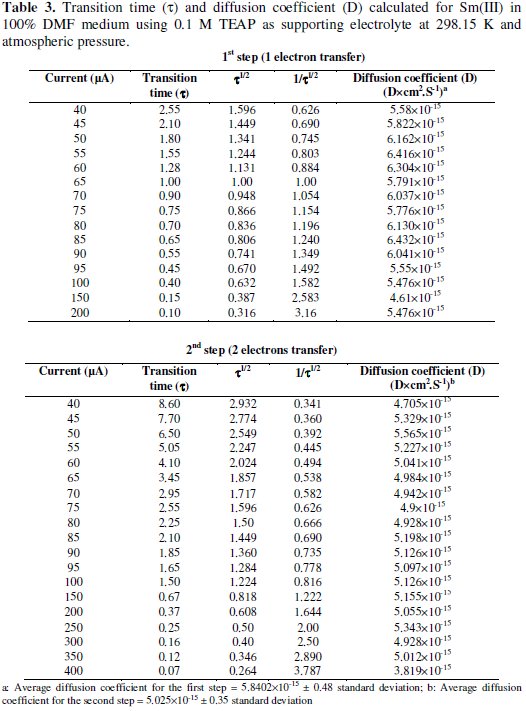
The linear behaviour of plot i vs. 1/β1/2 clearly suggests that the reductions of Sm(III) to Sm(II) and Sm(II) to Sm(0) were governed by diffusion process and not affected by other complications, as shown in Fig. 3.
The linear regression for the first step is y = 59.56 X + 6.332, and for the second step is y = 112.1 X + 7.774.
Chronopotentiometry of Pr(III) ion
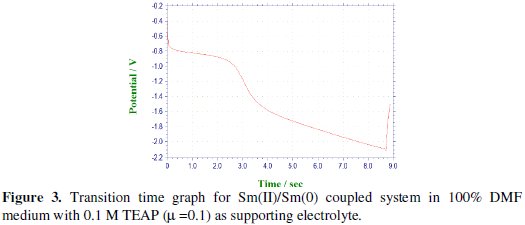
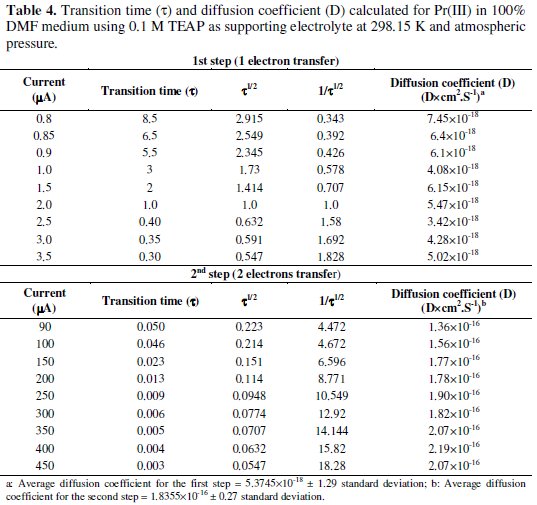
Under the specified experimental conditions, Pr(III) was investigated by cathodic process in 0.1 mol/L TEAP in 100% DMF medium. The cathodic segments of Pr(III) were recorded after adjusting the potential in the range of -0.2 to -1.0 V for the first step of reduction by varying the current density from 0.8 to 3.5 μA. Chronopotentiogram of Pr(III) in the cathodic process showed a single plateau with transition time (τ) 8.5 s at 0.8 μA current density, as depicted in Fig. 4, which was decreased with increase in the current density.
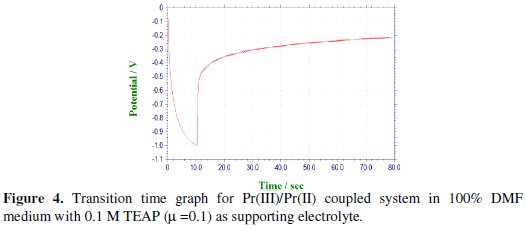
The cathodic segments for the second step were recorded after adjusting the potential in the range of -1.5 to -2.1 V by varying the current density from 90 to 450 μA, as shown in Fig. 5.
Chronopotentiogram of Pr(III) in the cathodic process showed a single plateau with transition time (τ) of 0.050 s at 90 μA current density, which was decreased with increase in the current density.
Beyond 450 μA current density, the shape of the curve was drawn out and hence, 450 μA was kept as the maximum limit for the second step of praseodymium. Chronopotentiometric data are summarized in Table 4.
Beyond 3.5 μA current density, the chronopotentiogram was found drawn out, hence 3.5 μA current density was chosen as the maximum limit. The linear behaviour of plot i vs. 1/β1/2 clearly suggests that the reduction of Pr(III) to Pr(II) and Pr(II) to Pr(0) was governed by diffusion process and not affected by other complications, as shown in Fig. 6.
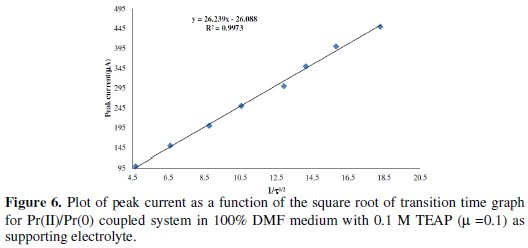
The linear regression for the first step is y = 1.543 X + 0.255, and for the second step it is y = 26.23 X - 26.08.
Conclusions
It was reported that out of 14 lanthanides only three of them, viz. europium, samarium and ytterbium, were known for their divalent state because of their electronic configurations. Very recently, the characterization of divalent state of europium and neodymium in non-aqueous medium was demonstrated using cyclic voltammetry and chronopotentiometric techniques [28]. In the present paper we have successfully characterized divalent state of Sm(III) and Pr(III) ion under specified experimental conditions in non-aqueous medium. To achieve this we have utilized potentiostatic and galvanostatic methods. Cyclic-voltammetry was used as potentiostatic method and chronopotentiometry was used as galvanostatic method. The calculated electrode kinetic parameters like diffusion coefficient and heterogeneous forward rate constant are found in good agreement with each other. Two distinct transition times (τ) from chronopotentiometry and two well-defined peaks in cyclic voltammetric method clearly suggest that under the specified experimental conditions and in non- aqueous medium both lanthanide ions exhibit in two steps. The first step is attributed to one electron transfer, the second step is attributed to two electrons transfer at the electrode surface, and this second step is dedicated for divalent states for samarium and praseodymium ions in non-aqueous medium.
References
1. Webb G A. Nuclear magnetic resonance. vol 34. Massachusetts. Athenaeum Press Ltd; 2005. [ Links ]
2. Homer RB, Mortimer BD. FEBS Lett. 1978;87:69. [ Links ]
3. Jelinek L, Arai T, Yuezhou W, et al. J Rare Earths. 2007;25:1. [ Links ]
4. Nipuni-Dhanesha HG, Yujiang M, Joel G, et al. Angew Chem Int Ed Engl. 2010;49:8923. [ Links ]
5. Joel G, Matthew JA. Eur J Inorg Chem. 2012;29:4550. [ Links ]
6. Straka M, Korenko M, Lisy F, et al. J Rare Earths. 2011;29:798. [ Links ]
7. Namy J L, Girard P, Kagan HB. New J Chem. 1977;1:5. [ Links ]
8. Kagan H B. J Alloys Comp. 2006;421:408. [ Links ]
9. Gareth J R. Tetrahedron. 2009;65:8603. [ Links ]
10. Michal S, Malcolm S, David JP. J Org Chem. 2012;77:3049. [ Links ]
11. David J P, Robert A F, Troels S. Organic Synthesis using Samarium Diiodide (SmI2). chap 1. London. The Royal Society of Chemistry; 2010. [ Links ]
12. Alexander S, Christoph S. Monat chemie. 2001;132:855. [ Links ]
13. Xue L, Yong-Min Z. J Zhejiang Univ Sci B. 2006;7:198. [ Links ]
14. Teprovich J A, PhD thesis, Lehigh University, Pennsylvania, USA, 2008. [ Links ]
15. Mohamadreza K B, Mahdi S, Seyed J A, et al. Nucl Med Commun. 2013;34:5. [ Links ]
16. Mohamadreza K B, Mahdi S, Seyed J A, et al. Annal Nucl Med. 2013;27:253. [ Links ]
17. Denis G, Deniard P, Gautron E, et al. Inorg Chem. 2008;47:4226. [ Links ]
18. Evans W J, Perotti J M, Brady J C, et al. J Am Chem Soc. 2003;125:5204. [ Links ]
19. Su F H, Chen W, Ding K, et al. J Phys Chem A. 2008;112:4772. [ Links ]
20. Hasegawa Y, Adachi T A, Tanaka A, et al. J Am Chem Soc. 2008;130:5710. [ Links ]
21. Bhatt P N, Pathak T V, Mehta J P, et al. J Rare Earth. 2006;24:653. [ Links ]
22. Mehta J P, Bhatt P N, Misra S N. Trans SAEST. 2004;39:46. [ Links ]
23. Chen F, Bian Z, Huang C. J Rare Earths. 2009;27:345. [ Links ]
24. Wang Y, Jin Ho, Deng S, et al. J. Rare Earths. 2011;29:388. [ Links ]
25. Geng J, Xiaogang Q. J Rare Earths. 2010;28:820. [ Links ]
26. Nief F. In: Gschneidner Jr K A, Bunzli J-C G, Pecharsky V K, editors. Handbook on the Physics and Chemistry of Rare Earths. chapter 247. Elsevier B. V. Publishers; 2010. [ Links ]
27. Evans W J. J Am Chem Soc. 2015;137:369. [ Links ]
28. Mehta J P, Pandya H M, Pandya K I. J Rare Earths. 2012;30:392. [ Links ]
Acknowledgement
The corresponding author is thankful to Department of Science and Technology, Govt. of India, for providing financial support to carry out our part of this work (SR/FTP/CS-12/05).
*Corresponding author. E-mail address: jpm1569@yahoo.co.in
Received 5 February 2015; accepted 15 June 2015

