










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkPortugaliae Electrochimica Acta
versão impressa ISSN 0872-1904
Port. Electrochim. Acta vol.38 no.1 Coimbra jan. 2020
https://doi.org/10.4152/pea.202001001
ARTIGOS
Experimental and Theoretical Studies for mild Steel Corrosion Inhibition in 1.0 M HCl by Two Stereoisomers of Benzothiazinone Derivatives
M. Sahranea, T. Ghailanea, M. Galaib, A. Souizia, Ebn Touhamib, R. Ghailanea and R. Touirb,c*
(a) Laboratory of Organic, Organometalic and Theoretical Synthesis. Faculté des Sciences; Université Ibn Tofail, Kenitra, Morocco
(b) Laboratory of Materials, Electrochemistry and Environment, Faculty of Sciences; Université Ibn Tofail, Kenitra, Morocco
(c) Regional Centre of Matters for Education and Formation (CRMEF), Avenue Allal Al Fassi, Madinat Al Irfane BP 6210 Rabat, Morocco
ABSTRACT
Mild steel corrosion inhibition in 1.0 M HCl, by two stereoisomers of 2-phenyl-benzothiazin-3-one (BHT1) and 3-phenyl-benzothiazin-2-one (BHT2), was researched using experimental and theoretical studies. The experimental results showed that the inhibition efficiency depends on the concentration and molecular structure of the investigated compounds, and it reached a maximum of 95 % and 96 %, at 10-4 M and 10-5 M of BHT1 and BTH2, respectively. This difference can be explained by the formation of hydrogen bonds, in BHT1 case. In addition, the polarization studies indicated that the above mentioned products acted as mixed type inhibitors. The molecular structure influence on the corrosion inhibition efficiency was theoretically investigated using DFT calculations. The structural and electronic parameters were determined, and showed good agreement with the experimental results.
Keywords: stereoisomers of benzothiazinone derivatives, corrosion inhibition, mild steel, electrochemical measurements, theoretical calculations.
Introduction
Generally, acidic solutions, which are often used in the chemical industry, cause deterioration of materials and their alloys. In order to protect these materials against corrosion by this media, the use of inhibitors is one of the proposed methods that have been widely applied. It was reported that the inhibitor efficiency not only depends on the inhibitor molecular structure, but also on the metallic surface nature and the electrolyte’s chemical composition [1- 3]. The investigation and application of new corrosion inhibitors holds great interest in the fields of corrosion research and industrial chemistry [4-6]. Most of these inhibitors contain nitrogen, sulphur and/or oxygen atoms, which may be adsorbed onto the metallic surface and block the active sites [7-9]. In addition, in our previous study [10], the inhibition efficiency of benzothiazinone derivatives, 2-aryl-benzothiazin-3-one with aryl = Ph-Cl, and 3-aryl-benzothiazin-2-one with aryl = Ph-CH3, on mild steel corrosion in acidic environments, was studied, and the results showed good agreement between experiments and theory.
The aim of this work was to study the inhibition efficiency of two new organic compounds, namely 2-phenyl-benzothiazine-3-one (BHT1) and 3-phenyl-benzothiazine-2-one (BHT2), synthesized and characterized by Souizi et al. [11], on the mild steel corrosion in 1.0 M HCl, using experimental and theoretical methods. The theoretical calculations have been performed in aqueous and gaseous phases, using DFT at B3LYP/6-311++G** theory level. The solvent effect was taken into account by using the polarized continuum model (PCM), and water as solvent. The local and global reactivity indices were calculated and discussed.
Experimental details
Material preparation and inhibitors
The electrolysis cell was a borosilicate glass (Pyrex®) cylinder closed by cap with three apertures. The chemical composition of the mild steel which was used as working electrode is summarized in table 1. Prior to each experiment, the electrode was abraded using emery paper up to 1500 grades, cleaned with ethanol, washed with distilled water, and finally dried in hot air. The platinum plate and the Ag/AgCl were used as counter and reference electrodes. All potentials are referred with respect to this last electrode.
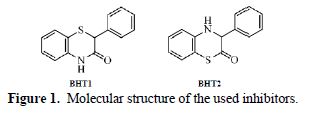
Electrochemical tests
The working electrode was immersed in a corrosive solution during 30 min, until the steady state of the open circuit potential (EOCP) was reached. The cathodic polarization curve was recorded by polarization from EOCP, towards more negative direction, with a sweep rate of 1 mV/s. After this scan, the same electrode was kept in the solution, until the open circuit potential steady state (EOCP ± 0.02 V) was obtained, and then, the anodic polarization curve was recorded from EOCP, to positive direction, with the same sweep rate. These measurements were carried out using PGZ 100 Potentiostat/Galvanostat monitored by a personal computer. For each concentration, three independent experiments were performed.
However, the overall current density (i) is considered as the sum of two contributions, anodic and cathodic current, ia and ic, respectively. For the potential domain, not too far from the open circuit, we can consider that both processes obey the Tafel law [12]; so, we can draw:
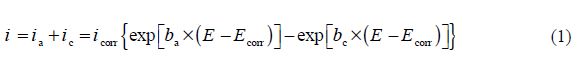
where icorr is the corrosion current density (A cm-2), and ba and bc are the Tafel constants of anodic and cathodic reactions (V-1), respectively. These constants are linked to the Tafel slopes β (V/dec) in a usual logarithmic scale given by the following equation:
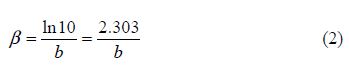
The corrosion parameters were then evaluated by means of the nonlinear least square method, applying equation (2), through Origin software. However, for this calculation, the applied potential range was limited to ±0.100 V around Ecorr; a significant systematic divergence was sometimes observed for both anodic and cathodic branches.
The corrosion inhibition efficiency was evaluated from the corrosion current density values, using the following relationship:
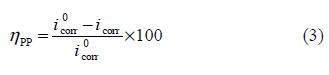
where and icorr are the corrosion current densities values, without and with inhibitor, respectively.
The electrochemical impedance spectroscopy measurements were carried out using a transfer function analyzer (VoltaLab PGZ 100), with a small amplitude a.c. signal (10 mV rms), over a frequency domain from 100 kHz to 100 mHz, with five points per decade. The EIS diagrams were done in the Nyquist representation. The experiments were repeated three times to ensure reproducibility. The results were then analyzed in terms of an equivalent electrical circuit, using Bouckamp program [13], and the inhibition efficiency was calculated using the following equation:
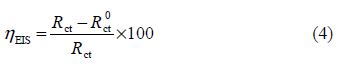
where and Rct are the charge transfer resistance values, with and without inhibitor, respectively.
Computational details
Full geometrical optimization of BHT1 and BHT2, in the gas and solution phases, was carried out using the Density Functional Theory (DFT), with the Beck’s three parameter exchange functional, and the Lee–Yang–Parr non local correlation functional (B3LYP) [14–16], combined to B3LYP/6-311++G** standard basis of atomic orbitals, as implemented in Gaussian 03 program package [17]. In addition, the physical or chemical properties measured in a solvent could be different, if they were measured in another solvent, or even in vacuum [18-21]. Moreover, it is well known that the phenomenon of electrochemical corrosion occurs in the liquid phase, where the inhibiting molecules show a behavior that is different to that without solvent. Consequently, it is necessary to include the solvent effect in the geometric and electrical properties calculations. In fact, the solvent effect on the molecular structures of the investigated compounds was considered, in the present work, by a model known as polarized continuum model (PCM) [22], implemented in Gaussian 03 program. In this model, the solvent was treated as a continuum dielectric media, and the solute was considered as a trapped molecule in a cavity surrounded by the solvent.
The geometry of the studied compounds was determined by optimizing all the geometric variables in the gas and aqueous phases, without any symmetry constraint, using analytical energy gradients. Harmonic frequencies were calculated from analytical derivatives for all species, to verify minimum energy structures. The obtained quantum chemical parameters from this optimized structure have been determined and analyzed, in order to explain the interaction between the inhibitor molecules and the metallic surface. According to DFT-Koopmans’s theorem [23–25], HOMO energy is related to the ionization potential (I), whereas LUMO energy is linked to the electron affinity (A), as follows:


Then, the electronegativity (c), the chemical potential (m) and the global hardness (h) were evaluated, based on the finite difference approximation, as linear combinations of the calculated I and A:


The fraction of transferred electrons (DN), evaluating the electronic flow in a reaction of two systems with different electronegativities (in this particular case, a metallic surface and inhibitor molecules), was calculated according to Pearson theory [26], as follows:
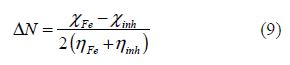
where the Fe and inh indices refer to the iron atom and the inhibitor molecule, respectively.
Results and discussion
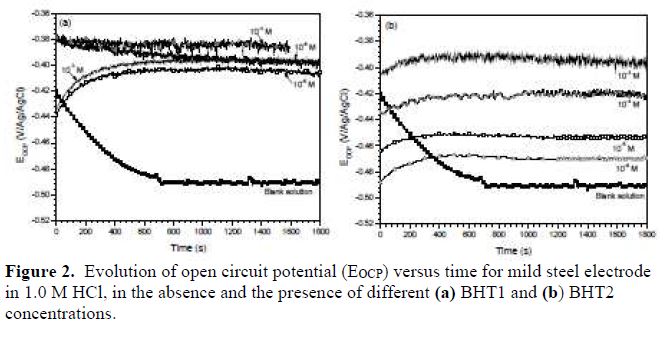
Evolution of open circuit potential (OCP) versus time
Fig. 2 shows the open circuit potential (OCP), versus time, of BHT1 and BHT2, at different concentrations, on the mild steel corrosion in 1.0 M HCl. It is noted that the potential for the solution without inhibitor gradually decreases with time, and stabilizes at the value of -490 mV/Ag/AgCl, after 12 min of immersion. This phenomenon characterizes mild steel corrosion with the formation of corrosion products. So, in the inhibitors presence, the potential values gradually increase with time towards the positive directions, and stabilize after almost 15 min of immersion. This finding indicated the formation of a protective layer at the metallic surface.
Potentiodynamic polarization curves
Fig. 3 presents BHT1 and BHT2 influence, at different concentrations, on mild steel corrosion in 1.0 M HCl. It is noted that the addition of these compounds decreased the current densities, and shifted the corrosion potential (Ecorr) towards positive values. Therefore, these inhibitors act as mixed type inhibitors.
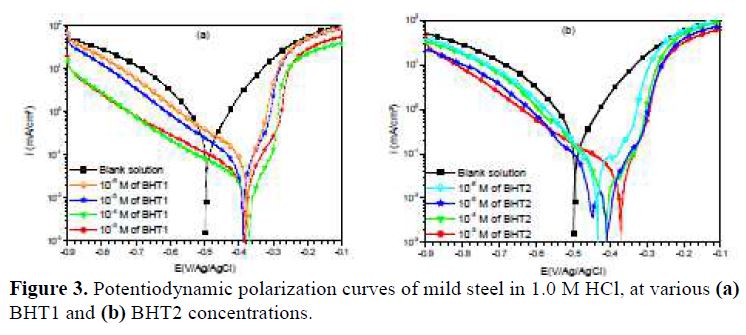
However, icorr, Ecorr, ba and bc were evaluated from the experimental results, using a defined function of “non-linear least squares curve fit” (Eq. (1)) of the graphic software (Origin, Origin Lab). In all cases, R2 correlation factor is greater than 0.99, indicating a reliable result. Fig. 4 shows, as an example, the results of regressions calculations for the cathodic and anodic branches, in the presence of different BHT1 concentrations.
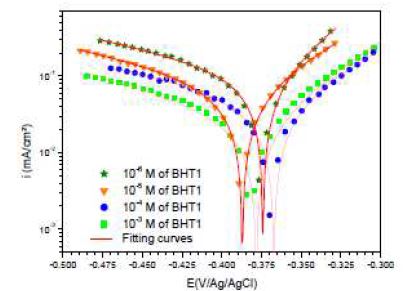
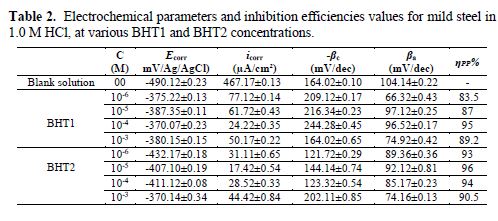
It can be seen, in this figure, good agreement between the experimental and fitting data. The obtained electrochemical parameters and the corresponding inhibition efficiencies (ηPP%) are listed in table 2. It is seen that the inhibition efficiency increased with the inhibitors concentrations, to attain the maximum of 94 % and 96 %, at 10-4 M of BHT1, and 10-5 M of BHT2, respectively. These results may be explained by the inhibitors molecules adsorption onto the mild steel surface, through non-bonding electron pairs present on nitrogen, oxygen and sulfur atoms, as well as π-electrons [27]. Thus, this variation mainly depends on the mutation of -N by -S atom. In addition, the low BHT1 inhibitive performance, compared to BHT2, can be explained by the formation of hydrogen bonds between –NH and =O, in BHT1 case. The same result was found and explained in our previous study, when we have studied the effect of monosaccharide for ordinary mild steel, in a simulated cooling water system [28].
Electrochemical impedance spectroscopy
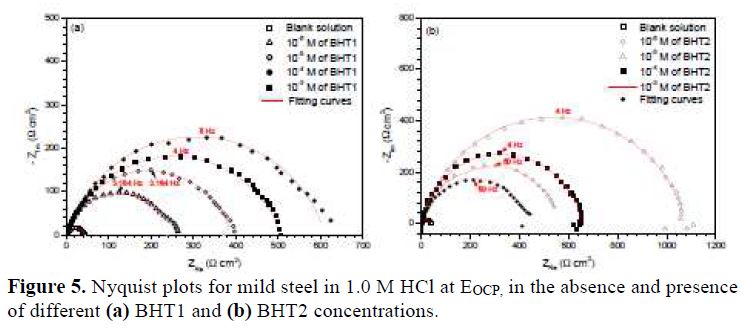
In order to describe the kinetics and characteristics of the electrochemical reactions occurring on the metal/electrolyte interface, the EIS analyses of mild steel in 1.0 M HCl, in the absence and presence of different BHT1 and BHT2 concentrations, at the open circuit potential (EOCP), were carried out.
Fig. 5 represents the obtained Nyquist plots. It is shown that the obtained semicircles are imperfect, resulting in the roughness and inhomogeneities of the metallic surface, which can cause the accumulation of corrosion products, and the formation of pits and cracks. This difference in the metallic surface state has been attributed to the frequency dispersion imputed to different physical phenomena [29].
A simple electrical equivalent circuit (Fig. 6) has been proposed for modeling the experimental data, where Rs represents the resistance electrolyte, and Rct is the charge transfer resistance. Qdl is the constant phase elements used to replace the double layer capacitances. Constant phase elements have been widely used to account for deviations brought about by surface roughness [30-31].
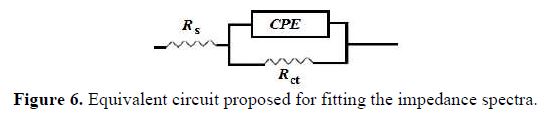
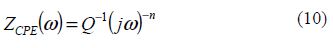
where Q is the constant phase element (CPE), ω is the sine wave modulation angular frequency (in rad s-1), j2 = -1 is the imaginary number and n is the CPE exponent. According to n value, CPE can be resistance [ZCPE = R, n = 0], pure capacity [ZCPE = C, n = 1], inductance [ZCPE = L, n = -1] or Warburg impedance (n = 0.5) [32].
However, the calculated double layer capacitance (Cdl) is derived from the CPE parameters, according to the following equation [33]:

and the relaxation time constant (τd) is derived from the charge-transfer process, using the following equation [33]:

The most important parameters derived from the fitting of the impedance diagrams are presented in table 3. It is seen that Rct values increase, whereas Cdl values decrease with BHT1 and BHT2 concentrations. These results can be attributed to a decrease in the local dielectric constant, and/or to an increase in the thickness of the electrical double layer, by the inhibitors adsorption [34]. It is also remarked that the inhibition efficiency increases with the inhibitors concentrations, to attain the maximum of 94 % and 96 %, at 10-4 M of BHT1 and 10-5 M of BHT2, respectively.
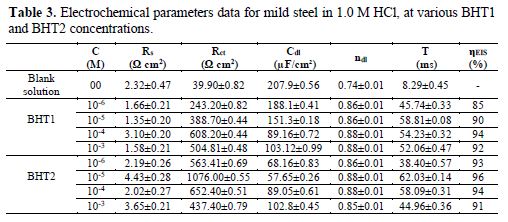
However, it is noted that ndl values are well below 1, and reflect the very flat shape of the impedance diagrams. So, the increase in ndl with the inhibitors concentrations reflects the reduction in heterogeneity of the mild steel surface, which results from BTH1 or BTH2 molecules adsorption onto the metallic surface [35-36]. In the same way, the relaxation time constant (τdl) values increase with the inhibitors concentrations; therefore, the adsorption process takes much more time, which means it is slower [37]. This showed that there is an agreement between the amount of charge that can be stored (that is, capacitance) and the discharge velocity in the interface (τdl) [33, 38]. In addition, the BHT2 charge transfer resistance is higher than that of BHT1, such as found by the potentiodynamic polarization curves.
In the other hand, in order to explain the experimental obtained results, quantum chemical parameters, such as highest occupied molecular orbital energy (EHOMO), lowest unoccupied molecular orbital energy (ELUMO), (LUMO–HOMO) energy gap (DE), dipole moment (m), electronegativity (c), ionization potential (I), electron affinity (A), global hardness (h) and fraction of electrons transfer (DN) from the inhibitor molecules to iron were determined and discussed.
Quantum chemical study
Molecular geometry
The molecules were built with the Gauss View 3.0 implemented in Gaussian 03 package [16]. Their corresponding geometries were fully optimized at B3LYP/6-311++G** level of theory, and the vibrational calculations prove that their equilibrium structures correspond to the minimum energy for each compound (absence of imaginary frequencies). Moreover, BHT1 and BHT2 geometries were re-optimized in the aqueous phase, at the same theory level, using PCM model for a better approach of the experimental parameters. The final geometries and the computational results parameters of the investigated compounds are given in Fig. 7 and table 4, respectively.
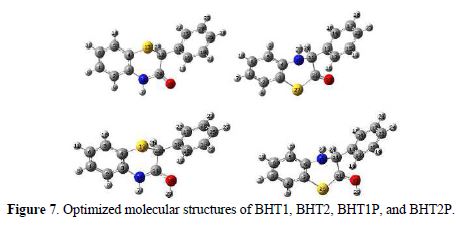
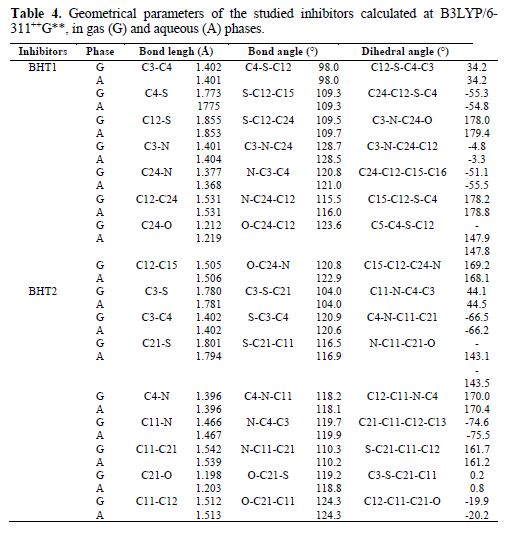
The comparison of BTH1 and BTH2 optimized geometrical structures in vacuum showed that the bond length C=O is longer in BHT1 than in BHT2, by about 0.014 Å, and that the C24-N bond in BHT1 is shorter than the single C11-N in BHT2, by about 0.089 Å. These differences can be explained by the delocalization in O=C24-N region, in BTH1. The C-C bond, where the first carbon is linked to the oxygen atom, and the second to S, in BHT1, and to N, in BHT2, showed a bond length difference of about 0.011 Å. In addition, large values of the bond lengths were observed for C-S; for the C21-S, the bond length, in BHT2, is shorter than C12-S, in BHT1, by 0.054 Å. From this, it can be concluded that the adsorption onto the metallic surface is clearly easier with C-S, for BHT2, than for BHT1.
However, the investigated compounds are not planar. This result was explained by the bond angles values of C4-S-C12 and S-C12-C15, which are about 98.0 and 109.3°, respectively, for BHT1, and 104.0° and 110.3°, for C3-S-C21 and N-C11-C21, respectively, in BHT2 case (table 4). The dihedral angle values of C24-C12-C15-C16, in BHT1, and C21-C11-C12-C13, in BHT2, exhibited an important distortion, by about 23.5°. In addition, a distortion, by about 10°, within the thiazine ring, was also observed by comparing dihedrals angles values of C12-S-C3-C4, in BTH1, and of C11-N-C4-C3, in BHT2. This result explains why the adsorption onto the metallic surface was easier for BHT2 than for BHT1.
On the other hand, it is noted that the solvent effect is slightly weak on the dihedrals and bonds angles, except for the C24-C12-C15-C16 dihedral angle in BHT1, which increases by about 4.4°; the greatest increase in bonds lengths (about 0.01 Å and 0.007 Å) was observed for C24-N, in BHT1, and for C21-S, in BHT2, respectively. This is probably due to the polarization of these bonds by the attraction of the carbonyl group. The bond length value of C=O increases by 0.007 Å, and 0.005 Å, in BHT1 and BHT2, respectively
Global molecular reactivity
Frontier orbital theory is frequently used to predict or confirm the inhibitor molecules adsorption centers responsible for the metallic surface/molecule interaction [39-41]. The terms involving the frontier molecular orbitals (FMO) could provide a relevant contribution, because of the inverse dependence of the stabilization energy on the orbital energy difference (DE = ELUMO - E HOMO). HOMO energy (EHOMO) is often associated to the electron donating ability of the molecules; therefore, inhibitors with high EHOMO values have a tendency to donate electrons to the appropriate acceptor, with low empty molecular orbital energy. Conversely, LUMO energy (ELUMO) indicates the molecules’ electron accepting ability; therefore, inhibitors with low ELUMO values have a tendency to accept electrons. The gap energy between the frontier orbitals (DE) is another important factor in describing the molecular activity, so, when the gap energy decreases, the inhibitor efficiency is improved [42]. Khaled has investigated the adsorption of thiourea and their derivatives on iron surface in nitric acid media, and he has found that the gap energy decreases with the increase in the inhibition efficiency [43].
The calculated quantum chemical parameters related to the inhibition efficiency of the studied molecules are shown in table 5. It is seen that the highest EHOMO was obtained for BHT2, which is not sufficient to conclude that it is more efficient than BHT1. Therefore, EHOMO high values are likely to indicate a tendency of the molecule to donate electrons to the appropriate acceptor molecules with low empty molecular orbital. Indeed, the excellent corrosion inhibitors are usually the organic compounds, which not only give electrons to the unoccupied orbital of metal, but also accept free electrons from it [39]. Thus, ELUMO and gap energy values obtained for BHT2 show that it has the highest inhibition efficiency. This result is in good agreement with the experimental efficiency - ηi (table 5).
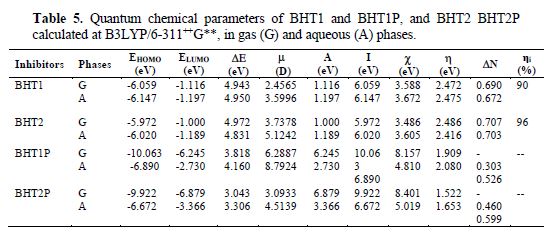
In addition, the dipole moment (µ) provides information on the polarity of the whole molecule. High dipole moment values are reported to facilitate the adsorption (and therefore, the inhibition), by influencing the transport process through the adsorbed layer [44]. Several authors have stated that the inhibition efficiency increases with dipole moments values [45-46]. On the other hand, literature survey reveals that many irregularities appear in the correlation of dipole moments with the inhibition efficiency [39, 47]. The dipole moments in the aqueous phase of BHT1 and BHT2 are 3.5996 D and 5.1242 D, respectively, which are higher than those of H2O (µ = 1.88 D). The high dipole moment value of these compounds probably indicates strong dipole-dipole interactions between them and the metallic surface [48]. Accordingly, the benzothiazine molecules adsorption in the aqueous solution can be regarded as a quasi-substitution process of the water molecules by the inhibitors molecules at the metallic surface (H2Oads). Experimentally, the corrosion inhibition study was made in an acidic solution; thus, to enrich the theoretical discussion, we considered the BHT1P and BHT2P protonated forms of BHT1 and BHT2, respectively. The protonation is promoted on the O atom, because the delocalization of non-bonding pairs of electrons from N and S atoms, in BHT1 and BHT2, respectively, occurs towards the O atom.
However, the fraction of electrons transferred (DN) from the inhibitor to the mild steel surface was also calculated using the theoretical values of cFe and hFe (cFe = 7 eV mol-1, and hFe = 0 eV mol-1) [49]. ΔN values are correlated to the inhibition efficiency resulting from electron donation. According to Lukovits et al., if ΔN < 3.6, the inhibition efficiency increases with an increased electron-donating ability at the metal surface [50]. The obtained ΔN values reported in table 5 showed that BHT2 has the highest ΔN value (0.703) in the aqueous phase. In addition, the highest inhibition efficiency experimentally obtained for BHT2 can be explained by the molecule tendency to receive the electron by the S atom in the unoccupied orbital (3d). This ability to receive the electron from the metallic surface increases the inhibition efficiency.
The calculated quantum chemical parameters in the solvent (water) presence and in the gas phase do not exhibit important differences (table 5). However, a slight modification has been observed for these parameters in the solution. Accordingly, it would be preferable to make computations in the gas phase, because it reduces the calculations cost, without a significant difference in the results.
From table 5, DE values of BHT1P and BHT2P are inferior to those found for BHT1 and BHT2; furthermore, it can also be clearly seen that the global hardness (h) of the protonated forms is inferior to that calculated for BHT1 and BHT2, which lets to predict their high reactivity. While the charge transfer (ΔN) taking place from the BHT1P and BHT2P inhibitor molecules to the metal is lower than that of BHT1 and BTH2 in the vacuum, as well as in the solution, this result can be explained by the fact that BHT1P and BHT2P, in the solution, can also receive the charge from the metal, which enhances the interaction metal-inhibitor. However, the lower ΔE value obtained for BHT2P, compared to that find for BHT1P, led us to conclude that the former is more efficient as an inhibitor than the latter.
The optimized geometries of the studied compounds in the neutral form, including their HOMO and LUMO distributions density, are shown in Fig. 8.
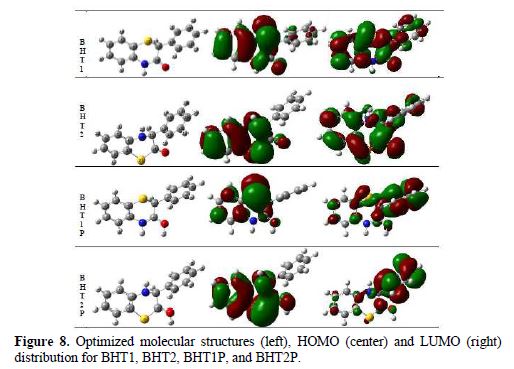
It can be seen that BHT1 and BHT2 have similar HOMO and LUMO distributions, which are all mainly located on the benzothiazin-3-one, in BHT1 moiety, and benzothiazin-2-one, in BHT2 moiety. These results indicate that the interaction between these molecules and the mild steel surface takes place on the benzothiazin-3-one part of BTH1 and BTH2.
As it can be seen from Fig. 8, most of LUMO distribution is observed on the phenyl group, for BHT1P and BHT2P; this distribution is more important for BHT2P.
On the other hand, HOMO distribution of BHT1P is essentially localized on the sulphur atom; this localization is less important on the C1, C3, C5 and O atoms of benzothiazinone. HOMO distribution of BHT2P is delocalized on the whole of the benzothiazinone part; this distribution is more significant than that observed for BHT1P. From these results, it can be deduced that BHT2P is promoted to interact with the metal, and to be an efficient inhibitor.
Local reactivity of the two benzothiazine derivatives
The local reactivity of BHT1 and BHT2 was investigated using the condensed Fukui indices (FI) [51]. Therefore, the molecules’ regions where the Fukui function is large are chemically softer than the regions where the Fukui function is small. The condensed Fukui functions [52] are found by taking the finite difference of approximated atomic charges, computed by natural population analysis (NPA), which was carried out in terms of localized electron-pair ‘bonding’ units [51].
The natural bond orbital (NBO) analysis [51] was performed for evaluating the electron-density distributions. So, for the neutral system of N electrons, independent single-point calculations were made at the same theory level than for corresponding N+1 and N-1 electron systems. In order to characterize the electronic population and the charge transfer capacity on each nuclear center, natural population analysis (NPA) [53] was done.
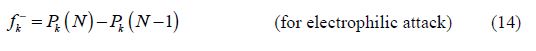
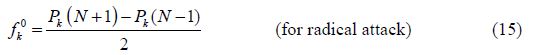
The calculated Fukui indices for all the charged species (N+1 and N-1), as well as for the neutral ones (N) of BHT1 and BHT2, are presented in table 6.
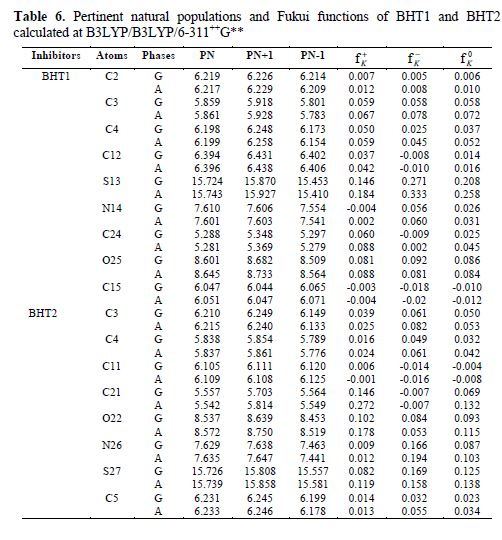
Local reactivity is analyzed by means of the condensed Fukui functions, in order to distinguish each part of the molecule on the basis of its distinct chemical behavior, due to the position of the -NH group in the thiazine ring. Therefore, the promoted sites for the electrophilic attack are C3, O and S for BHT1, and C3, C4, O, N and S for BHT2, while the favored sites for the nucleophilic attack are C3, C4, C24, O and S, for BHT1, and C21, O and S, for BHT2.
From table 6, it can be concluded that BHT1 and BHT2 have many active centers for adsorption onto the mild steel surface. Thus, the areas containing N, O and S atoms are the most favored sites for bonding with the mild steel surface through donating electrons. However, the S atom can give and receive electrons to and from the metal, respectively. The last process reinforces the inhibitor molecule adsorption onto the metallic surface, and both processes are more accentuated in BHT2 than in BHT1. This result confirms the experimental results, which showed, in BHT1 case, that the inhibition efficiency was decreased by the formation of a hydrogen bond.
Considering the solvent influence, it can be judged that there is not a remarkable difference between the gaseous and aqueous phases for the Fukui indices. The sites of nucleophilic and electrophilic attacks, in particular, for the S atom, are more pronounced for BHT2 than for BHT1 compounds.
Conclusion
The inhibition efficiency of mild steel corrosion in 1.0 M HCl, by two stereoisomers of benzothiazinone derivatives, has been investigated using electrochemical measurements and quantum chemical calculations. From the results it was concluded that:
- Potentiodynamic polarization curves indicated that both compounds act as mixed type inhibitors, and that their inhibition efficiency increases with their higher concentrations.
- The electrochemical impedance spectroscopy techniques confirmed the polarization results, and that BHT2 inhibition efficiency is greater than that obtained by BHT1, which can be explained by the formation of a hydrogen bond.
- The geometrical parameters showed that BHT2, because of its low distortion, is more efficient for the corrosion inhibition, in comparison to BHT1.
- The density distributions of the frontier molecular orbitals showed that BHT1 and BHT2 adsorb, through their active centers, nitrogen, oxygen, sulfur and π electrons of the benzothiazine ring.
- The Fukui indices proved that the reactive sites for the electrophilic and nucleophilic attacks of the BHT2 compound are the most suitable for adsorption.
- The theoretical results are in good agreement with the experimental ones.
References
- Alibakhshi E, Ramezanzadeh M, Bahlakeh G, et al. Journal of Molecular Liquids.2018; 255: 185–198.
- Gupta NK, Joshi PG, Srivastava V, et al. Chitosan. International Journal of Biological Macromolecules. 2018; 106: 704–711.
- 3. Salhi A, Tighadouini S, El-Massaoudi M, et al. Journal of Molecular Liquids. 2017; 248: 340–349.
- Yadav DK, Quraishi MA, Maiti B. Corros Sci. 2012; 50: 254–266.
- Yuce AO, Kardas G. Corros Sci. 2012; 58: 86–94.
- Zarrok H, Zarrouk A, Salghi R, et al. Int J Electrochem Sci. 2013; 8: 6014–6032.
- Donnelly B, Downie TC, Grzeskowiak R, et al. Corros Sci. 1977; 18: 109–116.
- Obot IB, Obi-Egbedi NO, Umoren SA. Corros Sci. 2009; 51: 276–282.
- Tadros AB, Abd El-Nabey BA. J Electroanal Chem. 1988; 246: 433–439.
- Ghailane T, Balkhmima RA, Ghailane R, et al. Corrosion Science. 2013; 76: 317–324
- Ghailane T, Saadouni M, Boukhris S, et al. Heterocycles. 2011; 83: 357–363.
- Stern M, Geary AL. Electrochemical polarization: a theoretical analysis of the shape of polarization curves. J Electrochem Soc. 1957; 104: 56–63
- Bouckamp A. Users Manual Equivalent Circuit, Ver. 4.51, 1993
- Becke AD. J Chem Phys. 1992; 96: 9489–9495.
- Lee C, Yang W, Parr RG. Phys Rev B. 1988; 37: 785–789.
- Becke AD. J Chem Phys. 1993; 98: 1372–1377.
- Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 03, Revision B.01, Gaussian, Inc., Pittsburgh, PA, 2003.
- Shaker MA, Abdel-Rahman HH. Am J Appl Sci. 2007; 4: 554–564.
- Shaker MA, Khalifa MA. Alex J Pharm Sci. 1995; 9: 159–161.
- Masoud MS, Ali AE, Shaker MA, et al. Spectrochim Acta A. 2004;60:3155–3159.
- Masoud MS, Ali AE, Shaker MA, M. et al. Spectrochim Acta A. 2005;61:3102–3107.
- Cances E, Mennucci B, Tomasi J. J Chem Phys. 1997;107:3032–3042.
- Hehre WJ, Radom L, Schleyer PvR, et al. Ab Initio Molecular Orbital Theory. 1986; Wiley-Interscience, New York.
- Janak JF. Phys Rev B. 1978;18:7165–7168.
- Stowasser R, Hoffmann R. J Am Chem Soc. 1999;121:3414–3420.
- Pearson RG. Inorg Chem. 1988;27:734–740.
- Bentiss F, Traisnel M, Lagrenée M. Appl Surf Sci. 2000;161:196-202.
- Touir R, Dkhireche N, Touhami ME, et al. Desalination. 2009;249:922-928.
- Mansfeld F. Corrosion. 1981;36:301-307.
- Felhosi I, Telegdi J, Palinkas G, et al. Electrochim Acta. 2002;47:2335-2340.
- Georges C, Rocca E, Steinmetz P. Electrochim Acta. 2008;53:4839-4845.
- Tsuchiya H, Fujimoto S, Chihara O, et al. Electrochimi Acta. 2002;47:4357-4366.
- Tazouti A, Galai M, Touir R, et al. Journal of Molecular Liquids. 2016;221:815–832
- Faustin M, Maciuk A, Salvin P, et al. Corros Sci. 2015;92:287-300.
- Popova A, Christov M, Vasilev A. Corros Sci. 2007;49:3290-3302.
- Azaroual MA, El Harrak EF, Touir R, et al. Journal of Molecular Liquids. 2016; 220:549-557.
- Rochdi A, Touir R, El Bakri M, et al. Journal of Environmental Chemical Engineering. 2015;3:233-242.
- Popova A, Christov M, Vasilev A. Corros Sci. 2011;53:1770-1777.
- Zhao P, Liang Q, Li Y. Appl Surf Sci. 2005;252:1596–1607.
- Fang J, Li J. J Mol Struct (Theochem). 2002;593:179–185.
- Bereket G, Hur E, Og?retir C. J Mol Struct (Theochem). 2002;578:79–88.
- Musa AY, Mohamad AB, Kadhum AAH, et al. J Indus Eng Chem. 2012;18:551–555.
- Khaled FK. Appl Surf Sci. 2010;256:6753–6763.
- Popova A, Christov M, Deligeorigiev T. Corrosion. 2003;59:756–764.
- Sahin M, Gece G, Karci F, et al. J Appl Electrochem. 2008;38:809–815.
- Quraishi MA, Sardar R. J Appl Electrochem. 2003;33:1163–1168.
- Khaled NF, Babic-Samardzija NK, Hackerman N. Electrochim Acta. 2005;50:2515–2520.
- Ramji K, Cairns DR, Rajeswari S. Appl Surf Sci. 2008;254:4483–4493.
- Obot IB, Obi-Egbedi NO. Corros Sci. 2010;52:657–660.
- Lukovits I, Kalman E, Zucchi F. Corrosion. 2001;57:3–9.
- Parr RG, Yang W. J Am Chem Soc. 1984;106:4049–4050.
- Yang W, Mortier WJ. J Am Chem Soc. 1986;108:5708–5711.
- Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899–926.
Received September 3, 2017; accepted April 4, 2018
* Corresponding author. E-mail address: Touir8@yahoo.fr; touir8@gmail.com

