











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Unmodified ethyleneamines form stable complexes with certain metal ions, such as zinc (Zn), nickel (Ni) and copper (Cu). This property makes them useful in many specialized applications, such as electroplating and electroless metal coating, as well as in the etching and stripping of Ni and of its alloy coatings. EDA, DETA and THPrED are used relatively often in galvanic processes. Both EDA and DETA play the role of Ni complexing agents, wherein the latter works more effectively that the former, as it is related to the complete Ni coordination only by two of its molecules, which allows the use of a lower complexant concentration [1]. THPrED is easily soluble in water, forming slightly alkaline solutions. Primarily, it is applied as a complexing agent for chemical Cu plating, and as a stabilizer in electrolyte solutions used in electroplating processes [2]. Furthermore, it is also used for the deposition of three- and multi-component galvanic coatings [3].
EDA oxidation has been mainly studied in organic solvents. A permanent modification of platine (Pt) and Au (gold) electrode surfaces, after the oxidation of pure neutral or acidic ethylenediamine (EDA), was observed by Harlem et al [4]. They found that Pt and Au were strongly coated with an organic compound through an oxygen-metal bond. The presence of water results in less stable formed layers.
A mechanism for EDA anodic polymerization was proposed. Initially, the formation of the NH2CH2CH2NH2+ radical cation occurs. It is followed by the C-N bond breaking, NH2 expulsion, and simultaneous formation of the primary NH2CH2CH2+ carbocation. This cation reacts with an NH2 group of an EDA molecule, yielding a new amine that can be further oxidized. The product on the anode is a polyethyleneimine-like polymer. A similar product was found for DETA, of which oxidation was also studied [5]. The mechanism leading to the binding of the amino group to the C, Au and Pt electrodes surface has been defined [6]. The analysis of the aliphatic amines through cyclic voltammetry (CV) indicates that the first intermediate is a radical cation that deprotonates, giving a radical along the sequential pathway. It is this radical that binds with the electrode surface. Therefore, the reaction path to the amines grafting should follow the reactions: RCH2NH2 -1e(( RCH2NH2 (( - H+( RC(HNH2(RCH2NH((GC(NHCH2R [6]. With water, DETA oxidation leads to the formation of imines, after solvated electrons release and deprotonation. Subsequently, imines are hydrolyzed to aldehydes and ammonium ions. Due to water decomposition, oxygen evolution takes place, which hinders DETA oxidation products adsorption onto the electrode surface [7].
Currently used methods for purifying water from galvanic processes containing amines are based on the chelated metal complexes decomposition into amines and metals, and the subsequent precipitation of metal ions. The most common practice for the chemical separation of metals in a chelated form involves a treatment with dithiocarbamate (DTC/CH2NS2), bisulphite (NHSO- 3), sulphide (S2−), iron salts, or their mixtures. These chemicals are often effective in lowering the concentration of various metals in the wastewater, enabling the achievement of the required levels. However, they are toxic, hazardous, harmful to the environment, and generate waste that may require further treatment to make it suitable for removal. Many papers point to the problems of various industries related to the treatment of wastewater containing complexed metals, especially THEtED, which are biologically highly stable and not easily degradable [8]. The literature identifies eighteen methods for separating metal ions from the wastewater, indicating their approaches and limitations [9].
The use of electrochemical oxidation technologies (EOTs) as an alternative to other classical advanced oxidation processes (AOPs) shows the high efficiency of the organic pollutants abatement, with significant advantages, such as versatility, energy efficiency, a relative easiness of automation and environmental compatibility, as the result of operating with a limited use of chemicals [10].
In recent years, much attention has been devoted to EO using anodes with a high over-potential of O2 evolution, such as BDD electrodes [11], which are excellent materials, with the highest electrochemical potential window, in aqueous solutions, from -1.5 V to + 2.5 V, generating hydroxyl radicals during that process. Enache et al. described the formation of these radicals, in a wide pH range from 1 to 11 [12]. BDD electrode has a high electrochemical performance, and electrochemically active species generally are not adsorbed onto its surface. As the literature reports, it can produce large amounts of highly reactive heterogeneous OH radicals on its surface, possessing high oxidation and mineralization power for the organic pollution treatment [13-16].
The main aim of this work was the detailed investigation of the selected ethyleneamines (particularly, DETA and THPrED) EOT by the BDD electrode. So far, there is no report on the EO of such compounds, in the terms of their disposal. DETA and THPrED degradation efficiency by the BDD electrode was evaluated under different current intensities and reaction times.
Materials and methods
Materials and reagents
The electrodes used in the experiments were: BDD/Nb, 2500 ppm, boron doping; BDD/Ta, 2500 ppm; BDD/Si, 700 ppm, 25 x 50 mm; BDD/Si, 700-800 ppm (as NeoCoat®-RDE Switzerland); and BDD/Si, 10 x 40 mm, with a boron doping concentration of ~ 3 x 1020 atoms/cm3 (Element Six Ireland). Purified water was used, with 18 MΩ resistivity (double purification, using the Millipore Milli-Q Plus system). DETA and THPrED purity levels were 99% and 98%, respectively (Sigma-Aldrich). EDA and methanesulfonic acid (MSA) purity was >99% and 70.0%, respectively, in water (Merck). Na2SO4 (sodium sulfate), NH4Cl (ammonium chloride), CH3COOK (potassium acetate) and acetone purity was 99%, 99%, 98% and 99.5%, respectively (Chempur, Poland). NaHCO3 (sodium bicarbonate), Na2CO3 (sodium carbonate) and standards for IC (Metrohm Poland) were used.
The test solution contained 1.91 g/L-1, i.e. 0.0185 mol/L-1 DETA, or 4.99 g/L-1, ie. 0.017065 mol/L-1 THPrED, in a 0.1 mol/L-1 Na2SO4 solution. The amine concentrations used are comparable to their contents in rinsed waters from galvanic processes.
Analytical measurements
To evaluate amines degradation, the following analytical methods were applied: total organic carbon (TOC) and total nitrogen (TN) were measured, using TOC-L, with a TNM (total nitrogen module -L analyzer (Shimadzu). Mineralization was calculated based on the following equations:
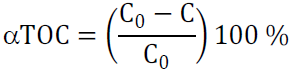 ***
***
where (TOC is TOC conversion (%), C0 and C are the organic carbon content, and TN0 and TN are the total nitrogen, in the solution (mg L-1), before and after the reaction, respectively. The samples were diluted 50 times with distilled water.
DETA and THPrED concentrations were analyzed employing IC, using a 930 Compact IC FIEX 250 Methorm apparatus equipped with a Metrosep C Supp 4-250/4.0 column. The mobile phase was an aqueous solution of MSA (15 x 10-3 mol/L-1) with acetone (5%). The mobile phase flow and the injection volume were 1 mL/min-1 and 20 μL, respectively. Anions were analyzed using a Metrosep A Supp 3 - 250/4.6 column, with the mobile phase consisting of acetone (50%) and water, with sodium bicarbonate (NaHCO₃) (4 x 10-3 mol/L-1) and sodium carbonate Na2CO₃ (1 x 10-3 mol/L-1).
Voltammetry experiments
Cyclic voltammetry (CV) and differential pulse (DP) voltammetry were used in the electrochemical experiments, with an Autolab PGSTAT-20 (Eco-Chemie, The Netherlands) potentiostat running with GPES 4.9 software. A three-electrode cell with a volume of 20 cm3 was used in all experiments. The BDD/Si disk (700-800 ppm) was used as an anode (electrode surface of 0.096cm2). A saturated calomel electrode (SCE) and a Pt mesh surrounding the anode were applied as reference and counter electrodes, respectively. DP and CV measurements were carried out using a BDD electrode with a geometric surface of 2.0 cm2. The measurement conditions were a pulse amplitude of 25 mV, a modulation time of 50 ms and a scan rate of 10 mV s-1.
Electrochemical degradation
Electrochemical degradation experiments were carried out in a cell with a volume of 50 cm3. The BDD/Si anode (Element Six Ireland), with a geometric surface area of 4.0 cm2, was used in all trials. A Pt mesh cathode was placed around the anode. Na2SO4 (0.1 mol/L-1) was used as supporting electrolyte (s.e.). Constant current (I) and reaction times (t), in the ranges from 0.4 to 0.9 A, and 0.5 to 3 h, respectively, were used during the amines oxidation reactions. Before each measurement, the electrode was rinsed in distilled water and polarized (0.1 Vs-1 from 0 to 2.5 Vs-1 vs. SCE) in the basic electrolyte solution, until stable CV characteristics were obtained.
Electrode preparation before CV measurements
The electrodes pretreatment was performed in a suitable s.e. (Na2SO4). The electrodes were washed in water with the Milipore System (18 MW). Rotating disk electrodes (RDE) were placed in s.e. for 10 s, with 1000 rpm disk rotation. Afterwards, the RDE were polarized at the potential of 2.5 V, during 60 s, without rotation, and were stabilized in the solution during 20 s, at 1000 rpm and 0.0 V, and, finally, for 10 s, without rotation. The same procedure was used in the cathodic polarization at -1.5V. The quality of the electrode preparation was verified based on CVs repeatedly obtained in the potential range from 0 to 2.5 V, in the s.e. After the pretreatment, the electrode was immediately used, and not allowed to dry or be significantly exposed to the atmosphere. RDE were used without rotation.
Results and discussion
Electrodes characterization
The BDD electrode surface morphology was characterized by a Scanning Electron Microscope (SEM). Fig. 1a shows a continuous and rough surface with visible sharp grains of 200-500 nm. Such a small crystal size enables a higher surface area, and increases the number of active sites where electrochemical reactions can occur.
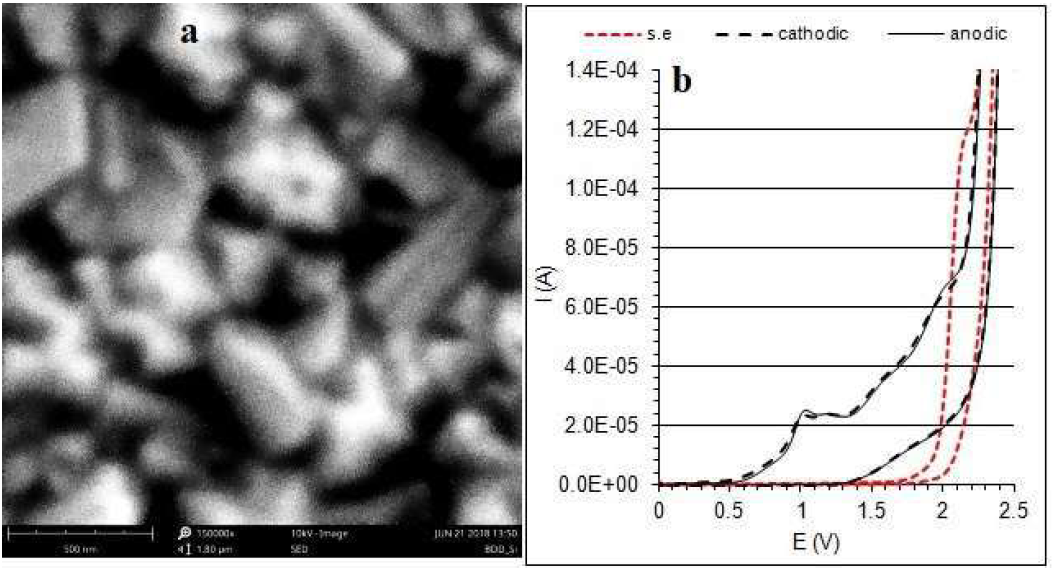
Figure 1 (a) SEM and SED (secondary electron detector) images (scale marker 500 nm) of the BDD/Si (700-800 ppm) electrode surface; (b) cyclic voltammograms of DETA oxidation after BDD/Si pretreatment by cathodic and anodic polarization (dashed line and solid line, respectively); s. e. (supporting electrolyte): 0.1 mol/L-1 Na2SO4.
The BDD electrodes were tested by cyclic voltammetry in 0.1 mol/L-1 H2SO4, Na2SO4 and NaOH solutions. It was found that the widest range of potentials for the cathodic and anodic reactions was obtained on BDD/Si electrodes, 700-800 ppm, 3 x 1020 atoms/cm3, in a Na2SO4 solution. Also under these conditions, a peak characterizing the formation of hydroxyl radicals was best shaped [12]. The direct evidence for •OH formation was obtained by the electron spin resonance method [17].
Depending on the anode material, the hydroxyl radicals can promote the partial and selective oxidation of the organic pollutant, and also its complete mineralization. To obtain reproducible results, the BDD electrode was previously electrochemically pretreated. The obtained CVs, in the potentials range from 0 to 2.5 V, in the s.e., were registered as an indicator of the electrode pretreatment quality. CVs of the DETA reaction (Fig. 1b) indicate that the electrode preparation method had no significant effect on the nature of the obtained curves. However, due to the investigation of the oxidation reaction, the anodic polarization (2.5 V) of the BDD electrode was performed for pretreatment in further measurements. This is important due to the slightly different oxidation profiles of the hydroxide ions in the anodic and cathodic pretreatment of the BDD electrode [18].
DETA and THPrED CVs
Preliminary information about DETA and THPrED electrochemistry was obtained from the current on the potential dependence. DETA (Fig. 2a) (0.0185 mol/L-1) and THPrED (Fig. 2b) (0.017065 mol/L-1) electrochemical behavior, in 0.1 mol/L-1 Na2SO4, was investigated by CV, at 100 mV s−1, over the potential range from 0.0 to 2.5 V.
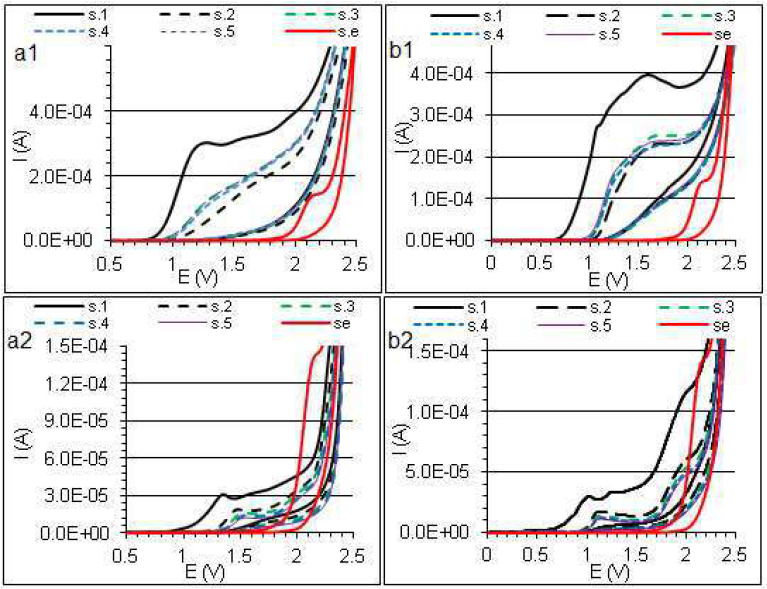
Figure 2 DETA (a1 and a2) and THPrED (b1 and b2) CVs in 0.1 mol/L-1 Na2SO4, at 25 ºC and 100 mV s-1, on the BDD disk electrode (0.096 cm2). s.1 and s.2 are the bolded solid and dashed lines scans, respectively, and s.3 to s.5 are the subsequent scans. (a1) 1.85 x 10-2 mol/L-1; (a2) 9.25 x 10-4 mol/L-1; (b1) 1.7067 x 10-2 mol/L-1; and (b2) 8.55 x 10-4 mol/L-1.
DETA oxidation peak occurred at the potential of 1.2 V, and also at higher potential values (Fig. 2a1), where CV curves did not overlap with the s.e. curve. Three poorly shaped peaks obtained at 10-fold DETA dilution confirm, at least, three steps of this reaction (Fig. 2a2). DETA oxidation reaction occurred irreversibly. A high decrease in the current between the first and the second scan (dashed line) and the wave shift is visible. In the next three scans, bad-shaped peaks shift in the opposite direction, with a simultaneous change in their currents, were observed. This indicates the polymer film formation on the BDD electrode surface, like on the other materials [5, 6]. Water presence results in less stable coatings [4, 6]. In the case of potentials higher than 2.0 V, the arising hydroxyl radicals caused further oxidation of the reaction products, and partial purification of the electrode.
Similar changes in the registered CV are noticed in the DETA solution with a concentration of 9.25 x 10-4 mol/L-1 (Fig. 2a2). However, in scans 3 and 4, at a potential of about 1.5 V, a shift in the peak current, to more positive values, is observed. Scan 5 is analogous to scan 4, which indicates that the reaction equilibrium has been established. The last wave, at a potential of about 2.0 V, was formed in the potential region where hydroxyl radicals were generated. The current of this wave is lower than that of the arising radicals (Fig. 2a1, s. e. curve), which indicates their lower formation. During THPrED oxidation (Fig. 2b), similar changes in the CV, as in DETA case, are observed. THPrED oxidation started at a lower potential than that of DETA.
Current intensity effect on the oxidation reaction
The changes in αTOC, αTN and αDETA, during 1 h of the electrochemical reaction, for different current intensities, are presented in Fig. 3a. This dependence proves that, after the oxidation reaction (1 h and 0.4 A), DETA practically disappears from the solution, while the removed TN value is about 20%. That indicates the formation of other products containing the amino group. The decrease in TOC is proportional to the increase in the current intensity, and αTOC = 95.485x + 0.5454, with R² = 0.9882.
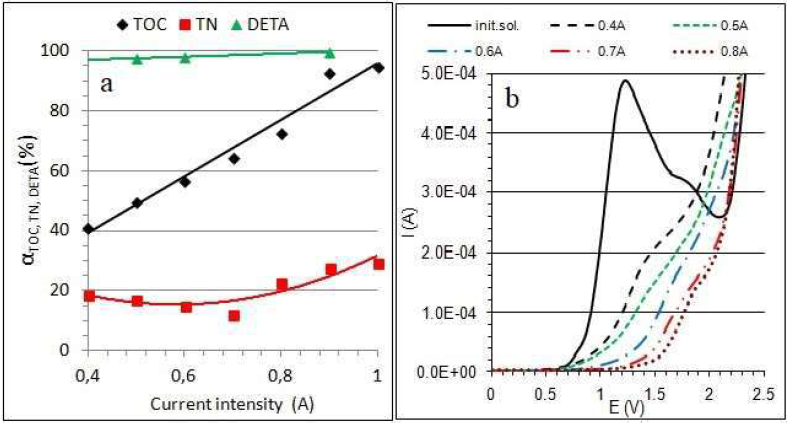
Figure 3 (a) Dependence of DETA decomposition degree, as αTOC, αTN and αDETA, on the current intensity, during 1 h of reaction; (b) DETA DPV: solid line - initial solution, dashed lines - after 1.0 h of electrolysis, at 0.4, 05, 0.6, 0.7 and 0.8 A, respectively.
To assess DETA conversion, DPV were also recorded. The measurements were carried out after the electrolysis, using the same electrode (geometric area of 2 cm2). In the initial solution, two peaks, for both the CV curve (Fig.2a) and the DPV curve (Fig. 3b), are observed. The first and the second arose at the potential of 1.2 V and around 1.7 V, respectively. However, after DETA oxidation reactions at different current values, the potential of poorly shaped DPV peaks shifted in the positive direction. It was found that the currents of these poorly-developed peaks are proportional to the TOC content, before and after DETA oxidation. Since the solutions after electrolysis practically do not contain DETA, the resulting peaks must characterize products of its oxidation. These products oxidize at higher potentials than those of DETA, and their DPV peaks shift from about 1.5 to 1.8 V.
Chromatographic analysis was carried out to determine the composition of the arising products. The best separation was obtained using IC. An exemplary chromatogram of the products formed in the reaction is shown in Fig. 4a. The peaks numbered as 1, 2, 3, 4, 5, 6 and 7 refer to the analysis on the cationic and anionic columns, respectively.
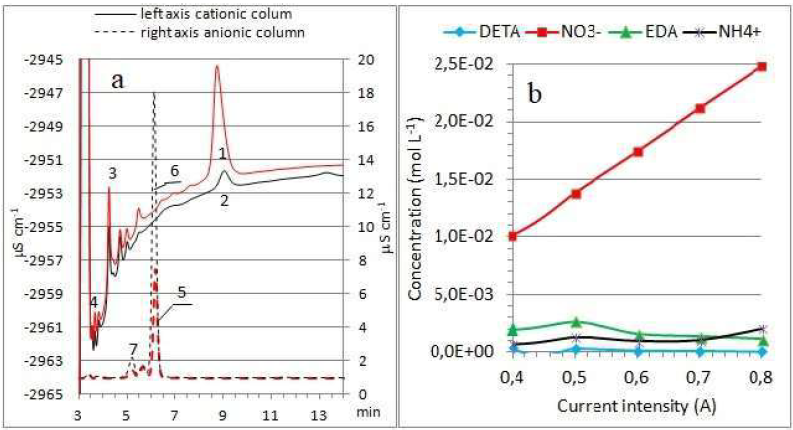
Figure 4 (a) Ion chromatograms of solutions after DETA oxidation (0.4 A) on the cationic and anionic columns. Peaks 1 and 2 - DETA, after 0.5 and 1 h of reaction, respectively; peak 3 - EDA; peak 4 - NH4 +; peaks 5 and 6 - NO3 (, after 0.5 and 1 h of reaction, respectively; and peak 7 - CH3-COOH. (b) Products concentration of DETA oxidation, with different current intensities, at 1 h of reaction.
Ouattara et al. have shown that, in non-aqueous solvents, with water, amines oxidation leads to the formation of NH4 + and aldehydes, as a result of the imine hydrolysis [7], and of a new amine which can be further oxidized [5]. In the aqueous environment, with hydroxyl radicals, acids and NO3- can also be formed. The changes in the concentration of reaction products with the increase in current are shown in Fig. 4b. The relationships show that NO3- ions are the main product of DETA oxidation. At a current from 0.4 to 0.5 A, an increase in EDA concentration is noticeable, while, at higher currents, its content decreases.
The THPrED oxidation reaction, in comparison with that of DETA, is slightly slower with increasing current intensities. During the one-hour reaction, at a current from 0.5 to 0.6 A, about 20% of the amine remain in the solution, whereas TN decrease is practically negligible (Fig. 5). TOC removal is proportional to the current intensity and αTOC = 80.386 x -0.0649, with R² = 0.9823.
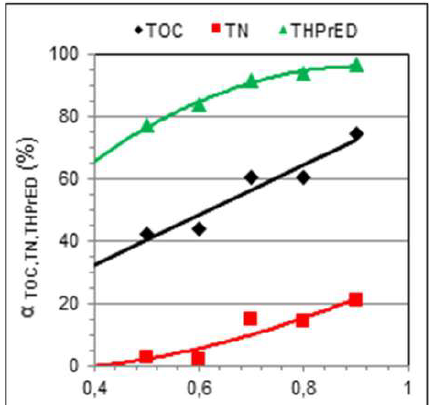
Figure 5 Dependence of THPrED decomposition degree, as αTOC, αTN and αTHPrED, on the current intensity, during 1 h of reaction. Current intensity
As previously, the composition of THPrED oxidation products was analyzed by IC. Chromatograms are shown in Fig. 6a.
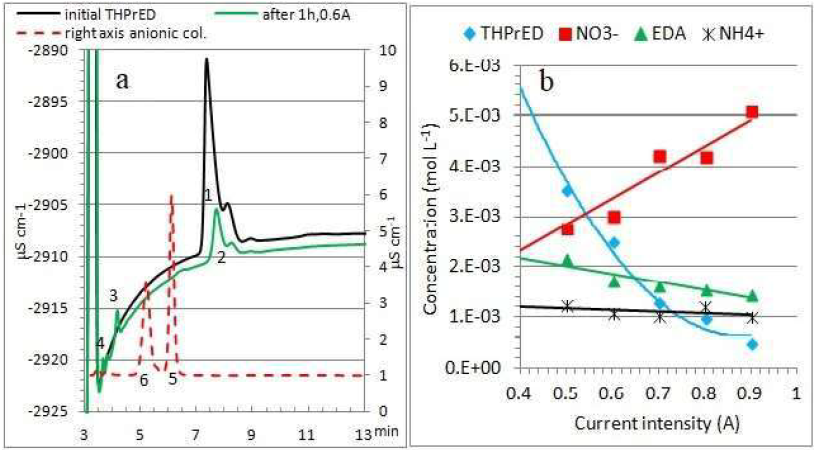
Figure 6 (a) Solutions IC after THPrED oxidation (0.6 A and 1 h) on cationic and anionic columns: peak 1 - THPrED initial concentration; peak 2 - THPrED oxidation; peak 3 - EDA; peak 4 - NH4 +; peak 5 - NO3 ( ; and peak 6 - CH3-COOH. (b) Products concentration of THPrED oxidation, with different current intensities, at 1 h of reaction.
The peaks numbered as 1, 2, 3 and 4, and 5 and 6 refer to the analysis of the cationic and anionic columns, respectively.
The changes in the concentration of reaction products with the current increase are shown in Fig. 6b. As follows from the presented dependencies, the resulting NO3- is the main product of THPrED oxidation. An increase in EDA, NO3- and NH4 + concentrations is observed only at a current intensity of 0.5 A. A further increase in current causes a decrease in their content.
CH3-COOH concentration increases with higher current intensities of DETA and THPrED oxidation. For currents higher than 0.6 A, CH3-COOH concentration decreases. During THPrED oxidation, CH3-COOH concentration is about two times higher than during DETA oxidation, at the current range used (Fig. 7).
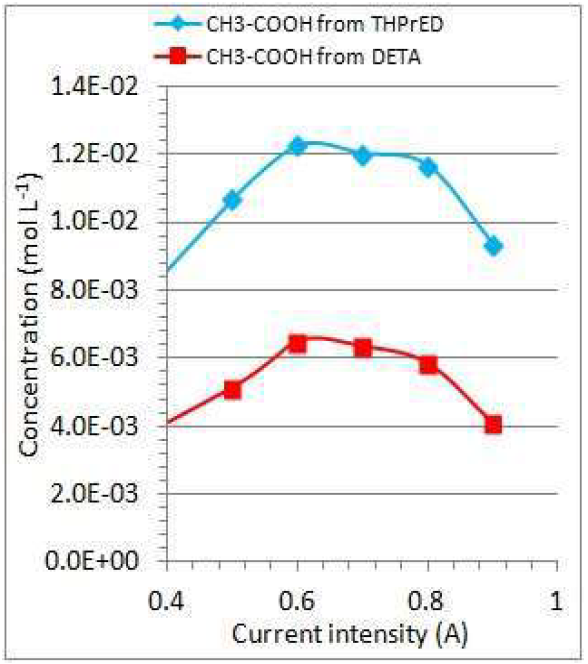
Figure 7 Dependence of CH3-COOH concentration changes on the current intensity, during 1 h of THPrED and DETA oxidation.
A comparison of the oxidation products for both substrates, at 0.6A, leads to the following statements:
Firstly, during THPrED oxidation, six times less of NO3- is formed, and, simultaneously, 10 times more of amine remain in the solution than in the case of DETA oxidation. Secondly, the TN content practically does not change after THPrED reaction, while it decreases at about 18% for DETA. This indicates partial destruction of the THPrED molecule, without elimination of the -N-C-C-N- bonds (EDA is one of the reaction products). It can be proposed that, during the DETA oxidation, one or two molecules of aldehyde are formed [7,19]. Next, these molecules are oxidized to acid. THPrED oxidation, by the breaking of the C-C bond, can lead to the formation of two or four alcohol molecules, which also can undergo further oxidation to acid.
The effect of time on the oxidation reaction
The current intensities of 0.4 A, for DETA, and 0.6 A, for THPrED, were used to determine the time effect on the amines oxidation. The changes in TOC removal, presented in Fig. 8a, are similar for both ethyleneamines.
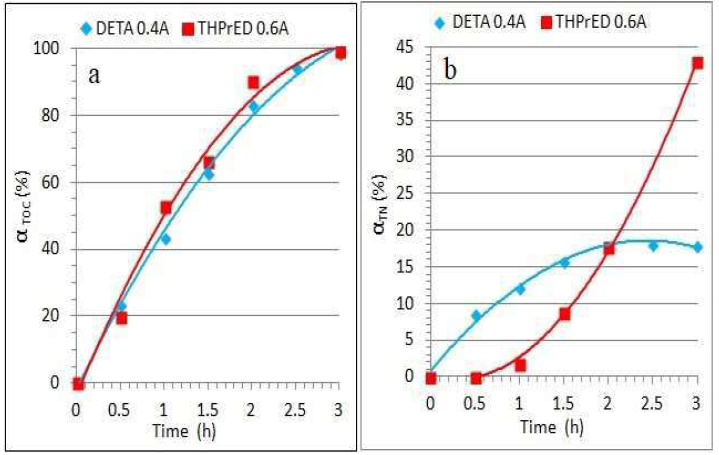
Figure 8 DETA and THPrED decomposition degree at current intensities of 0.4 and 0.6 A, respectively, with reaction times. (a) αTOC; (b) αTN.
In both solutions, an almost linear increase in TOC, up to about 90%, is observed. Two ranges of increase in αTOC can be distinguished: a relatively sharp one, within the time range from 0.5 to 2 h, and another much milder, in the range from 2 to 3 h. This means that the complete removal of the remaining 10% TOC extended the oxidation time by about 30%. Simultaneously, with TOC, TN content was analyzed (Fig. 8b). After 1.5 h of DETA oxidation, the decrease in TN was about 18%, but no further increase in αTN, with time, was observed.
TN removal, in THPrED oxidation case, was completely different. After 1 h of reaction, αTN reached just 2%, but during successive 2 h, a significant increase in it, up to more than 40%, took place. The concentration of products formed during the amines oxidation was calculated based on the chromatograms depicted in Figs. 4a and 6a. The changes in DETA concentration with time, at the current intensity of 0.4 A, are shown in Fig. 9a.
The above dependencies indicate that comparable amounts of products (0.4 x 10-2 mol/L-1) are present in the solution, if conditions such as 0.5 h and 0.4 A are applied. More precisely, their concentrations in mol/L-1 are as follows: DETA - 0.4268 x 10-2, EDA - 0.3652 x 10-2, NO3 - 0.3993 x 10-2 and NH4 + -0.04 x 10 - 2. Under such conditions, 8.6% of TN was also removed from the solution. Not all peaks registered on the chromatograms were identified. Therefore, changes in the peaks area for different current intensities of DETA oxidation were analyzed. The obtained relationships (not shown here) indicate a continuous decrease in the content of products formed during the reaction with the current increase.
Analogously, the dependence of THPrED oxidation products concentration on the reaction time (at 0.6 A) is presented in Fig. 9b. Despite the use of a higher current for THPrED oxidation, the concentrations of its reaction products are lower than those of DETA oxidation at a lower current. It should be noted that the oxidation time was the same in both cases.
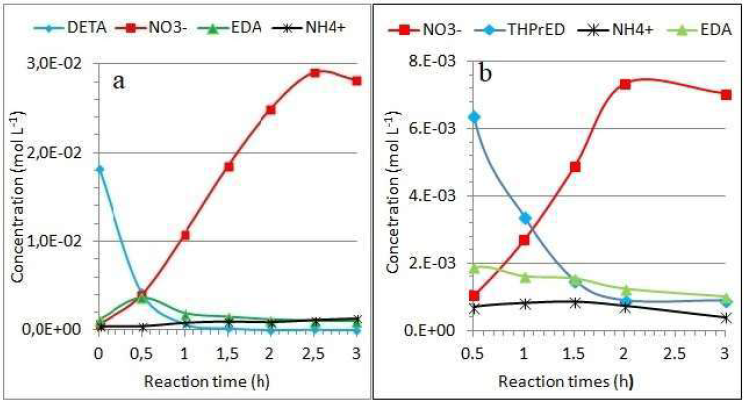
Figure 9 Changes in (a) DETA and (b) THPrED oxidation products concentration with reaction time, at current intensities of 0.4 A and 0.6 A, respectively.
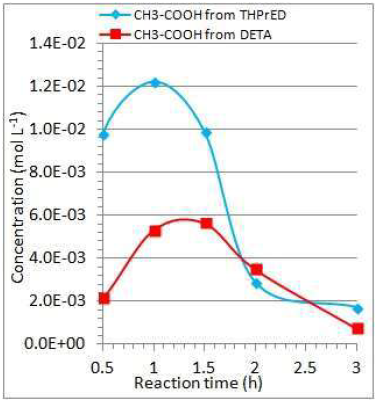
Figure 10 Dependence of acetate ions concentration changes, during THPrED and DETA oxidation, on reaction times, at a current intensity of 0.6 A and 0.4 A, respectively.
During the oxidation of both substrates, acetates were formed. Maximum concentrations were obtained after about 1 h of reaction. Approximately two times higher acetate concentration was found during THPrED oxidation than during DETA oxidation (Fig.10). The acetate concentration decreased about six fold as the oxidation time increased to 3 h, at the applied current intensity.
Analysis of proposed and probable reactions
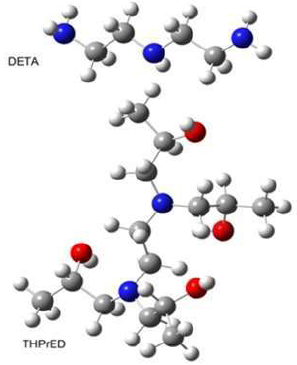
To make it easier to follow the DETA and THPrED decomposition pathways presented in this section, Fig. 11 recalls the structures of these compounds.
On the BDD electrode, in addition to electrochemical reactions, substrates and intermediates are oxidized by the formed hydroxyl radicals. The first reaction is the electrochemical discharge of water leading to hydroxyl radicals formation: BDD +H2O(BDD((OH) +H+ +e(.
These hydroxyl radicals are then consumed by two competing reactions, organic compounds oxidation and oxygen evolution: BDD((OH) +organics (BDD +CO2 +H2O +inorganic ions; BDD((OH) (BDD+1/2O2 +H+ +e(.
Based on the obtained results and literature data, the following scheme of electrochemical degradation for the analyzed compounds is proposed: both substrates partially block the BDD electrode surface. That seems to occur when the polyamine possesses two primary amines at both sides in the molecule structure [5]. The simultaneous oxygen evolution and formation of hydroxyl radicals hinder the adsorption of DETA oxidation products. DETA oxidation leads to the formation of an unstable imine, which is further hydrolyzed to aldehyde and ammonium ion [7]: NH2-CH2-CH2-NH-CH2-CH2-NH2 - e (NH2-CH2-CH2-NH-CH2-CH2-+(NH2; NH2-CH2-CH2-NH-CH2-CH2-+(NH2 - H+( NH2-CH2-CH2-NH-CH2-(CH-+(NH2; NH2-CH2-CH2-NH-CH2-(CH-+(NH2(NH2 -CH2-CH2-NH-CH2-CH ( NH2 (; NH2 -CH2-CH2-NH-CH2-CH ( NH2 ( + H2O ( NH2 -CH2-CH2-NH-CH2-CH ( O + NH4 +.
The resulting aldehydes are oxidized to acids that have been identified in the reaction products.
During the oxidation (0.5 A and 1 h), almost complete DETA destruction (98%), as well as the decrease in TOC by 50%, and in TN by 18%, were found. The main products of DETA removal were EDA, NH4 +, NO3, CH3-COOH, N2 and CO2. With the increase in reaction time, the concentration of the analyzed components decreased, except for NO3-, which is demonstrated by a constant TN. This indicates the total mineralization of the substrate to CO2, NO3- and H2O. After 180 min of reaction, αTOC and αTN values for DETA were 94% and 18%, respectively.
DETA (- e( and + (OH) ( reaction products were: R - CH ( O +NH4 + CH3-COOH + N2 + EDA+ CO2 + NO3 ( + H2O.
During THPrED oxidation (0.5 A and 1 h), its 80% destruction, about 40% decrease in TOC content, and practically no change in TN content, were found. After electrolysis, EDA, NH4 +, NO3- and CH3-COOH were identified in the solution. This indicates partial destruction of the THPrED molecule without total removal of the -N-C-C-N- bonds (EDA was one of the reaction products). Thus, the beginning of THPrED oxidation may be connected with the break of the C-C bond, resulting in the formation of two or four alcohol molecules. Alcohol undergoes further oxidation to acid, identified in the post-reaction solution. With the increase in the reaction time, the concentration of the analyzed components decreased, except that of NO3-ions. After 1.5 h of electro oxidation, an increase in TN removal and a practically unchanging amount of NO3- were observed. At the same time, the acetate content dropped significantly. The αTOC and αN final values reached 98.6% and 43.6%, respectively, after 180 min of reaction.
Conclusions
DETA and THPrED effective electrochemical removal using a BDD electrode proved to be a very promising method of potential practical application to wastewater treatment containing amines. It was found that nitrates, ammonium and acetates were formed in a short reaction time, and at low currents.
For DETA, at a constant reaction time (60 min), the organic carbon removal rate increased almost linearly with higher current intensities. During DETA reaction, from 30 to120 min, at a constant current, a linear increase in TOC and N conversion rate of was also observed. For longer reaction times (more than 120 min), TOC removal rate decreases, while N conversion rate slightly increased. After 180 min of reaction, αTOC and αN were 94.0% and 18%, respectively.
For THPrED, αTOC was equal to 98.6%, and αN was 43.6%. These values suggest that amines oxidation occurred not only on the BDD electrode, but also by hydroxyl radicals formed at the potentials of about 2 V and higher.

