











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntroduction
Many experimental methods and modern surface characterization tools have been created to evaluate and characterize the performance of corrosion inhibitors for metals and alloys 1-4. These methodologies are often expensive, time consuming and tedious. Computational methods have already proven to be very useful in determining the inhibitors molecular structure and elucidating its electronic properties and reactivity.
The aim of assessing the efficiency of a corrosion inhibitor, with the help of computational chemistry tools, is to search for compounds with desired properties, using mathematical quantified and computerized forms 5.
The development of DFT, force fields and molecular dynamic (MD) simulations, abolition, semi-empirical and Hartree-Fock model approaches provides potential solutions.
The pioneering work of Hohenberg and Kohn (1964) 6 and Kohn and Sham (1965) 7 has made DFT one of the more popular tools among computational chemical methods, because it focuses on the electron density P(r) as the carrier of all information in the molecular ground state, rather than on a single-electron wave function 1. Several molecular parameters and descriptors widely used in the molecular characterization of corrosion inhibitors effectiveness on the metal surfaces have been derived from DFT.
The purpose of our review is to divulge computational chemistry as a modern tool in developing novel molecules for the protection of metals and alloys that are important engineering contributors, and to examine papers and short communications that focus on this research topic. Investigation efforts that have been done recently on the use of computational tools in the field of corrosion studies are presented below.
Computational modeling in corrosion inhibition
Based in the frontier molecular orbital (FMO) theory description, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are often associated with the ability of inhibitor molecules to donate and accept electrons, respectively.
HOMO energy (EHOMO) indicates that an inhibitor molecule has the tendency to donate electrons to the metals unoccupied molecular orbitals (UMO).
In its turn, LUMO energy (ELUMO) indicates the ability of the inhibitor molecules to accept electrons from the metals occupied molecular orbitals (OMO). High ELUMO values mean that the inhibitor molecule accepts electrons more easily, and, if that happens, it is not considered as a good corrosion protector, since it will not tend to prevent metals deterioration. The principle behind this is that a molecule ELUMO must be lower than its EHOMO, for excellent corrosion inhibition 8.
Computational methods
The following computational methods, including Ab initio, DFT and semi-empirical models, are used for solving the time independent Schrödinger equation for the electrons of a molecular system, as a function of their nuclei positions.
The simplest type of Ab initio electronic structure calculation is the H-F, which does not take instantaneous coulomb electron-electron repulsion into account, and only included its average effect on its calculation 5. DFT is used to investigate the electronic structure, mainly the ground state of many-body systems, in particular atoms and molecules, and its condensed phase. Its main goal is to replace the many-body electronic wave function with the electronic density, as the basic quantity 9. Semi-empirical molecular orbital methods have been widely used in computational research, and neglect many smaller integrals, in order to speed up calculations. In current practice, they are efficient computational tools that can yield fast quantitative estimates for a given number of properties. This is particularly useful to correlate a large set of experimental and theoretical data 5.
Literature survey
Comparative data showing the significance of computational tools in the performance of some organic corrosion inhibitors on metals and alloys have been previously reported 8,50-64, and are shown in Table 1.
Table 1 Comparative chart showing the corrosion inhibitors performance on metal and alloys, using theoretical and experimental approaches.
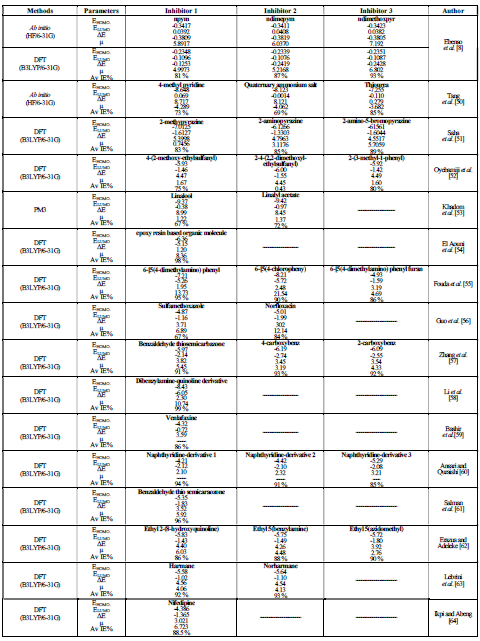
It can be observed that the molecules ELUMO is lower than their EHOMO, and that ΔE and μ values are within the range of those for excellent corrosion inhibitors.
Corrosion inhibition studies by DFT and Ab initio methods
Ozcan et al.10 theoretically explored the interactions between methionine molecules and a metal surface, by using the B3LYP/6-311G (d, p) method. The Fukui function index A, which is a local type of reactivity criterion, was considered, and Fukui indices were determined for N+1 and N-1 electron species, at the geometry of the methionic molecule reference N-electron. Their results showed that the FMO (HOMO and LUMO) localization and the condensed Fukui functions (f- and f+) analysis in the reactive region were very useful to characterize organic adsorbates.
Zarrouk et al. 11 investigated some quinoline derivatives, namely 1, 4-dihydroquinoxaline-2,3-dione(Q1) and 2-phenylthieno(2,3-b] quinoxaline (Q2), as corrosion inhibitors for mild steel (MS), using the B3LYP/6-31G DFT. The calculated values of quantum chemical parameters, such as EHOMO, ELUMO, energy gap (ΔE), dipole moment ((), electronegativity (X), electron affinity (A), global hardness (n), softness (σ), ionization potential (IP), fraction of transferred electrons (ΔN), global electrophilicity (w) and energy total (Etot) were correlated with their experimental results.
The Ab initio method with HF/LANL2DZ basis set was used by Hongfang et al. 12, to investigate the theoretical parameters of the inhibition property of three nitrogen-heterocyclic compounds, namely 3,5-dimethy-1H-pyrazole, pyridine and 2-(3-methyl-H-Pyrazole-5-yl) pyridine against steel corrosion 12. Their results revealed a good relationship between the corrosion IE and EHOMO. They also found that the lower the value of the combined energy, the higher the stability of the formed complex, confirming the inhibitor good corrosion protection property.
Rodríguez-Valdez et al. 13 investigated the electronic and molecular structures of several heterocyclic compounds, namely 1, 3, 4-thiadiazole and its derivatives, using the B3LYP/STO-3G basis set. Etot, ΔE, EHOMO and ELUMO, μ and η parameters were all calculated for each of the derivatives, both in the liquid and gas phases, of which results show no significant difference on the heterocyclic compounds structures.
Feng et al. 14 investigated adsorbate-surface interactions using the B3LYP/6-31G basis set method. They carried out some calculations of a self-assembled monolayer of 2-mercapto-benzothiazole (MBT), to obtain the following parameters: atomic charges, EHOMO and ELUMO and the interaction energy between MBT and iron (Fe). Benzothiazole results were compared with those of other studied molecules. The two MBTs EHOMO (-624.1 kJ/mol-1 for thiol and -583.5 kJ/mol-1 for thione) were larger than those of benzothiazole (-547 kJ/mol-1). These data indicate the significance of the exocyclic sulphur (S) atom that informs the self-assembled monolayers on Fe. The calculated interaction energy also revealed that MBT was chemically adsorbed onto the Fe surface, most likely through the exocyclic S atom.
Ab initio and a modified DFT called PBEg, with 3-21G, CBSB2, CBSB7, CBSB4 and CBSB1 basis sets, were used by Rodríguez-Valdez et al. 15, to study 4-amino-1, 2, 3, 5-thiatriazole and 5-amino-1, 2, 3, 4-thiatriazole compounds. A comparison between the results of this study and of previous computational simulations was made, since there were no experimental data for the researched compounds. A good agreement was found between those findings, which means that precise data could be obtained by using the modified DFT.
Rodríguez-Valdez et al. 16 also reported a similar study on the effectiveness of hydroxyethyl, aminoethyl and amidoethyl, using the PBEg functional with the CBSB2 basis set.
Hasanov et al. 17 studied the inhibitory performance of two Schiff base compounds, 2-[(4-Methoxy-phenylimino)-methyl]-phenol and 1-[(4-methoxyphenyl)imino]methyl-2-naphthol, on steel corrosion in a sulfuric acid (H2SO4) solution. Frontier orbital energies, Mulliken charges of possible adsorption sites and ( parameters were calculated using a B3LYP/6-31G(d) basis set. The theoretical data were found to be in good agreement with the experimental results.
Ju et al. 18 reported the theoretical study of the electronic and molecular structures of aminic nitrogen-containing polydenate Schiff’s base compounds using a B3LYP/6-31G basis set. EHOMO and ELUMO, charge distribution, absolute X values and the fraction of electrons transferred (ΔN) from the inhibitor to Fe were calculated. The results show that the inhibition efficiency (IE) increased in EHOMO and decreased in ΔE.
Yan et al. 19 presented purines and their derivatives, guanine, adenine, 2,6-diaminopurine, 6-thioguanine and 2, 6-dithiopurine, as corrosion inhibitors for MS in a 1 M HCl solution, using the 6-31GDFT method. The Mulliken partial charges, EHOMO, EHOMO, ΔE and ( were calculated, and the IE was associated with ΔE and the frontier orbital electron density. It was observed that purines were physically adsorbed onto MS.
Imidazole, benzimidazole and their derivatives IE was investigated by Zhang et al. 20, using the B3LYP/6-31G method. They calculated the number of quantum chemical parameters, such as EHOMO, ELUMO, ΔE, charge distribution, ( and molecular connectivity index (MCI), showing the molecule steric hindrance effect. The results revealed that EHOMO was the most statistically significant term influencing the corrosion IEs: the greater the EHOMO, the greater the IE.
FMO energies, ΔE, MO densities and ( of newly synthesized compounds, such as triazole derivatives, 4-chloro-acetophenone-O-(1,3,3-triazolyl)-metheneoxime (CATM), 4-fluoro-acetophenone-O-1-(1,3,4-triazolyl)-metheneoxime (FATM) and 4-methoxyl-aceto-phenone-O-1-(1,3,4-triazoyl)-metheneoxime (MATM), were investigated by Li et al. 21 as corrosion inhibitors for MS in acidic media, using Ab initio method and a 3-21G basis set. They were unable to find a direct relationship between the molecules IE and EHOMO, ELUMO and (. Rather, they reduced the adsorption process complexity by employing the free energy of adsorption (ΔGads) values obtained from the thermodynamic consideration. Based on ΔGads and on the calculated quantum chemical parameters, they observed two types of interactions, chemisorption and physisorption, as being responsible for the thetriazole derivatives inhibition behavior.
Ebenso et al. 8 investigated four rhodanine azosulpha drugs (5-sulfadiazineazo-3-phenyl-2-thioxo-4-thiazolidinone, 5-sulfamethazineazo-3-phenyl-2-thioxo-4-thiazolidinone, 5-sulfadimethoxineazo-3-phenyl-2-thioxo-4-thiazolidinone and 5-sulfamethoxazoleazo-3-phenyl-2-thioxo-4-thiazolidinone), as corrosion inhibitors for MS in an acidic medium, using DFT at the B3LYP/6-31G (d, p) and B3LYP/6-311G (d, p) basis set levels. Ab initio calculations using the HF/6-31G (d, p) and HF/6-311G (d, p) methods for quantum chemical parameters/descriptors, namely, EHOMO, ELUMO, ΔE, μ, A, I, X, η, (, polarizability (α), Mulliken charges and fraction of electrons transferred (ΔN) from the inhibitors to Fe, were calculated and correlated with the experimental IE%. Quantitative structure activity relationship (QSAR) approach and a composite index of some quantum chemical parameters/descriptors were also considered to characterize the studied molecules IE. The results showed that the IE% of the studied rhodanine azosulfa drugs was closely related to their theoretical IE%, but with varying degrees of the correlation coefficient (R2). The IE% also increased in EHOMO and decreased in EHOMO-ELUMO, and the areas containing N and S atoms were the most possible sites for bonding to the Fe surface, by donating the lone pair electrons to its empty d-orbital.
Corrosion inhibition studies by semi-empirical methods
The molecular structure effect on the inhibitor efficiency was studied by Al-Sabagh et al. 22, using H-F and PM3 (Parameterized Model no 3) level of theories. The electronic properties, such as EHOMO, ELUMO, μ and molecular orbital densities, were correlated with experimental data, and the results were in good agreement. Khaled and Amin 23 computationally and experimentally investigated the adsorption and corrosion inhibition behavior of four selected piperidine derivatives for nickel (Ni) in a 1.0 M HNO3 solution, using MD simulation, quantum chemical calculations and electrochemical Tafel and impedance methods. The MD simulation was performed using commercial software material studio, while quantum chemical calculations based on the Ab initio method were also performed to determine the relationship between the piperidine derivatives molecular structures and their IE. Results obtained from Tafel and impedance methods were in line with the data of the theoretical studies.
The corrosion inhibition effect of some imidazole derivatives on aluminum (Al) in hydrochloric acid (HCl) was also examined by Khaled and Amin 24. They used the Monte Carlo simulation technique, incorporating molecular mechanisms and dynamics to simulate the imidazole derivatives adsorption. Results of both techniques confirmed the derivatives good inhibitive action against Al corrosion.
Khaled 25 recently conducted a research on molecular simulation tools to optimize adsorbed triazole derivatives (aminotriazole and benzotriazole) structure. The Fe /inhibitor/solvent interface was simulated, and the charges on the inhibitor molecules, as well as their structural parameters, were calculated, considering the solvent effects. According to the experimental and theoretical data, aminotriazole was found to be better than benzotriazole.
Cardoso et al. 26-28 made a detailed experimental and theoretical investigation on 23 different compounds, including amines, thiourea derivatives and acetylenicalcohols, as corrosion inhibitors for 22% Cr stainless steel (austenitic-ferritic, duplex) in HCl solutions, employing AM1 (Austin model 1) and the quantitative structure-property relationship (QSPR). The AM1 method was used for calculating the quantum chemical parameters. Their results show that the experimental data correlated well with the theoretical information.
Other compounds, such as zinc di-alkyl-di-thiophosphates 29,30, potassium ethyl xanthate 31, phthalocyanines 32, polymethylene amines 33, benzyl triphenylphosphonium bromide 34, hydrazine carbodithioic acid derivatives 35, aliphatic amines 36, pyrimidine derivatives 37, piperazine derivatives 38, amino acids and hydroxy carboxylic acids 39, phenyl-N,N-dimorpholinemethanes 40, aniline trimers 41, triblock copolymers 42, quarternary ammonium salts 43, organophosphorus compounds 44, para-chlorobenzene nitriles 45, maleimide and its derivatives 46, anhydrides and imides 47, and substituted catechols and pyrogallols 48, were also investigated, using both experimental and semi empirical methods. Although there is no general way of predicting the potential of the studied compounds to be good corrosion inhibitors, their electron structural parameters have been extensively correlated with their corrosion IEs.
Based on this theory, attempts were made by Babic´-Samardzija et al. 49 to correlate some molecular parameters of N-heterocyclic amines (piperidine (pip), 2-methyl piperidine (2mp), 3-methyl piperidine (3mp), 2,6- dimethyl piperidine (26dp) and 3,5-dimethyl piperidine (35dp)) with their corrosion IEs, using PM3, AM1 and MNDO (modified neglect of differential overlap) semi-empirical methods. The compounds molecular structure effect on their inhibiting properties has been considered in terms of their electronic and chemical structures.
These researchers firstly studied the effect of electron density, which is the charge on the nitrogen atom and on the entire heterocyclic ring. Secondly, they investigated the effect of structural changes in terms of bond distances and angles. The Etot was obtained after the nuclear coordinates geometric optimization, according to the results of their computational study. A a relationship between amino compounds molecular parameters and their inhibition properties was found. Although this relationship is not so simple and straightforward as it might be expected, a number of neglected parameters that could be involved in it, such as surface and solution characteristics, could give, at least, a simplified explanation. It is also clear that these N-heterocyclic amines inhibition behavior could be due to the charge on the nitrogen atom, and to the sum of the net charge of the six atoms from the cyclic ring 49.
In general, any measurable property of an atomic or molecular system can be derived from the Schrodinger equation. The semi-empirical molecular orbital methods have been widely used in computational chemistry studies throughout the past decades. In order to speed up the calculations, semi-empirical methods have been used, ignoring several smaller integrals. Empirical parameters are inserted into the remaining integrals, in order to compensate for the errors caused by these approximations, and are measured against accurate experimental or theoretical reference results. This strategy can only be effective, if the semi-empirical model maintains the essential physics, to describe the properties of interest. The parameterization will account for all other effects in an average context, and assess the numerical accuracy of a given method will validate it. Semi-empirical methods are useful statistical instruments in current practice, producing quick quantitative estimates for various properties 66. This can be especially useful in comparing large sets of experimental and theoretical data, classifying patterns in similar molecular groups, and solving a computational problem, before continuing with higher-level methods.
With regard to the semi-empirical methods precision and applicability breadth, there is a need to improve them, without compromising their computational efficiency. Moreover, is necessary to create new algorithms, in order to explore modern computer architectures, and expand semi-empirical calculations for larger molecules. A large number of semi-empirical methods with different acronyms has been developed over the years, including AM1 67, MNDO 68 and PM3 69.
Concerning the approximations details (e.g. the core-core repulsion functions) and, especially, the parameters values, the semi-empirical methods differ. For various purposes, semi-empirical approaches can be optimized. MNDO, AM1 and PM3 methods have been developed to reproduce the formation of several organic molecules heat and structure. Other semi-empirical methods for studying spectroscopic properties include INDO/S (intermediate neglect of differential overlap) or CNDO/S (complete neglect of differential overlap ), and are precisely optimized.
MNDO
MNDO depends on the NDDO (neglect of the diatomic differential overlap) approximation, which is an improved version of the INDO method. INDO itself is an improvement of the CNDO approximation. There are several such semi-empirical LCAO-MO (linear combination of atomic orbitals) methods, developed for specific purposes 66.
AM1
AM1 is a semi-empirical method that depends on the NDDO integral. Specifically, it is a generalization of the modified NDDO approximation. AM1 was developed by Michael Dewar and coworkers, as reported in 1985 67.
AM1 attempts to improve the MNDO model, by reducing atoms repulsion at close separation distances. The atomic core terms in the MNDO equations were modified through the addition of off-center attractive and repulsive Gaussian functions. The parameterization problem complexity was higher in AM1, as its number of parameters per atom increased from 7, in MNDO, to 13-6.
PM3
PM3 was developed by Stewart, and first reported in 1989 68. It is another semi-empirical method based on the NDDO integral approximation.
The only differences between PM3 and AM1 are: PM3 uses two Gaussian functions for the core repulsion function, instead of the varied number used by AM1 (between one and four Gaussians per element); the parameters numerical values are different; and whereas PM3 treats some of the parameter values as optimized, AM1 takes them from spectroscopic measurements.
Mathematical methods ΔE
In quantum chemical studies, ΔE is an important computational parameter used for the prediction of the corrosion IE. It is a theoretical model that explains the structure and confirmation barrier of many molecular systems. Equation (1) is used to calculate ΔE 65.
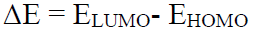 ***
***
IP and EA
IP is the maximum energy needed to remove an electron from many electron atoms in a gas phase (equation 2), whereas EA is the energy released when an electron is attached to a gas phase atom (equation 3).
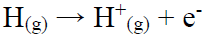 ***
***
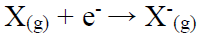 ***
***
The IP and EA were calculated using Equations 4 and 5.
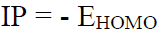 ***
***
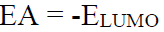 ***
***
Absolute electronegativity (()
( shows how atoms can attract electrons towards them. The Koopman’s theorem is always used in the estimation of a compound (, according to (equation 6).
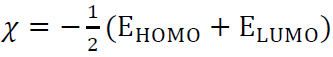 ***
***
The higher the χ value , the more effective is the inhibitor, and vice versa.
ƞ and σ
The measurement of an atom resistance to charge transfer is considered by ƞ, while σ describes the capacity of an atom or group of atoms to receive electrons (8, 65). These parameters are computed with the following equations:
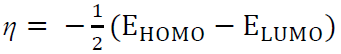 ***
***
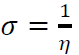 ***
***
Generally, a hard molecule and a soft molecule require large and small ΔEs, respectively. That means soft molecules can easily offer electrons to an acceptor system that makes them more reactive than hard molecules. Besides, adsorption may likely occur at the point of a molecule where σ is high 55.
μ
The μ measure of polarity in a bond, which involves the distribution of electrons in a molecule, is a powerful index frequently used to predict the corrosion inhibition process. Inhibitors with high μ values form strong dipole-dipole interactions with the metal, showing a strong adsorption onto its metal surface, which leads to higher IEs 65.
w index
High and low electrophilicity index values reflect a good electrophile and nucleophile, respectively. The relationship between electrophilicity and chemical hardness gives global electrophilicity index ((), according to the equation 9.
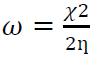 ***
***
ΔN can be evaluated using the following equation:
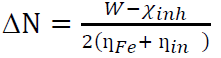 ***
***
where W is the work function, χinh is the inhibitor electronegativity, while ƞFe and ƞinh are Fe and the inhibitor global hardness, respectively. The work function is always given as W = 4.82, for the Fe (110) surface.
When the value of ΔN> 0, the electron transfer takes place between an inhibitor molecule and the metal surface, and when the value of ΔN < 3.6, the electron donating ability of an inhibitor molecule is increased.
Atomic charges
It is clear that electric charges in the molecule are responsible for electrostatic interactions. In several chemical reactions, and for the compounds physico-chemical properties, the local electron densities or charges are significant. In order to determine chemical reactivity indices of weak intermolecular interactions, charge-based parameters have been employed.
The definition of a partial atomic charge is somewhat subjective, despite its utility, because it depends on the method used to delimit one atom from the next.
There are several methods for calculating the partial charges.
Mulliken population analysis 69 is primarily used to measure the distribution of the charge in a molecule.
This method facilitates obtaining these numerical quantities and offers a qualitative understanding of the molecules structure and reactivity 70.
In addition, for the explanation of the molecular polarity, atomic charges are used.
Mulliken charges are used to locate the corrosion inhibitor molecules adsorption centers. High negatively charge atoms have a greater tendency to be adsorbed onto the metal surface 71-79.
Conclusions
The literature survey of computational chemistry studies is shown in Table 1, which reveals that computational chemistry software is a powerful tool to investigate the fundamental molecular level process for metals and alloys corrosion inhibition.
The role of computational chemistry study is to focus on the reduction of corrosion control costs.

