











 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
Permalink
Introdução
A microangiopatia trombótica é uma lesão das arteríolas e capilares que leva à trombose intraluminal, podendo evoluir com obstrução parcial ou total da microvasculatura. Clinicamente, apresenta-se com trombocitopenia e anemia hemolítica microangiopática. Este grupo de patologias tem como as suas duas principais entidades, a púrpura trombocitopénica trombótica (caracterizada pela deficiência na atividade da ADAMTS13) e a síndrome hemolítica-urémica típica (SHUt), seguindo-se as menos frequentes SHU mediada pelo complemento (SHUc), microangiopatia mediado por fármacos, por alterações na coagulação e no metabolismo, entre outras.1,2
A SHUt e a SHUc apresentam por sua vez características distintas e que levam a diferentes orientações terapêuticas. A SHUt, o mais comum, causado pela toxina Shiga secretada pela Escherichia coli (cujo serotipo mais comum é E. coli O157:H7) e a SHUc, mais rara (2/1 000 000 adultos na Europa), que pode ocorrer em qualquer idade sendo mais comum em crianças e jovens adultos, pode ser esporádica ou familiar (em menos de 20% dos casos) e que é causada por defeitos na regulação da cascata do complemento com subsequente ativação descontrolada da via alternativa.3,4No entanto estas alterações não estão sempre presentes.
Estão descritos vários triggers para o aparecimento da SHUc, tais como: infeções pelo vírus da imunodeficiência humana, infeções bacterianas por Mycoplasma pneumoniae e Streptococcus pneumoniae, gravidez, neoplasia, fármacos como tacrolimus, ciclosporina, cisplatina, clopidogrel, entre outros.1,3Tratando-se de causas menos frequentes, estes triggers podem também apresentar-se como causas secundárias de anemia hemolítica microangiopática.3 A apresentação clássica destas síndromes inclui anemia hemolítica, teste Coombs negativo, trombocitopenia e lesão renal aguda. Estão descritas manifestações extra-renais em 20%-30% dos doentes, maioritariamente do trato digestivo (25%), sistema neurológico, cardiopulmonar e oftalmológico.1,3A abordagem terapêutica nesta patologia foi durante muitos anos a permuta plasmática. Atualmente dispomos do eculizumab, anticorpo monoclonal com afinidade para C5, dirigindo o tratamento com o bloqueio da microangiopatia trombótica mediada pelo complemento.2,5,6O prognóstico na era prévia ao surgimento do eculizumab, era desfavorável em cerca de 50% dos casos, com evolução para doença renal terminal (DRT) e apresentava uma mortalidade de 25% em fase aguda.5
Caso Clínico
Apresenta-se o caso de uma doente de 59 anos, nacionalidade americana e com antecedentes de asma na infância, mononucleose infeciosa aos 16 anos, meningite vírica aos 22 anos e neoplasia do ovário submetida a histerectomia e ooforectomia aos 46 anos.
A doente recorreu ao serviço de urgência por quadro de náuseas, vómitos, dor abdominal difusa, dejeções líquidas com muco (mais de 5 dejeções diárias com pequena quantidade de sangue associado) e colúria com 3 dias de evolução. Ao exame objetivo apresentava pele e mucosas ictéricas, TA 115/65 mmHg, FC 65 bpm, temperatura 36,8ºC, saturações periféricas de 97% em ar ambiente, dor abdominal difusa à palpação, sem organomegalias palpáveis e exame neurológico sem alterações. Do estudo realizado salienta-se bicitopenia (anemia normocítica normocrómica com hemoglobina 8,3 g/dL e trombocitopenia 8000/uL), lesão renal aguda KDIGO 2 (creatinina 1,52 g/dL; ureia 74 g/dL) e hiperbilirrubinemia 9,5 mg/dL à custa da bilirrubina indireta, lactato desidrogenase 1430 U/L, haptoglobina <30 md/dL, exame sumário de urina com hematoproteinúria, teste de Coombs negativo e esfregaço sangue periférico com esquizócitos.
Realizou ecografia abdominal que excluiu alterações de relevo. A abordagem terapêutica inicial incluiu a transfusão de um pool de plaquetas, a administração de 500 mg de metilprednisolona, sendo posteriormente admitida no Serviço de Medicina Intensiva nível II (SMI) pela necessidade de realização de permuta plasmática.
À admissão no SMI a doente apresentava-se com Coma Glasgow Scale de 15, estável do ponto de vista tensional, sem necessidade de oxigénio suplementar e apirética, mucosas francamente ictéricas, com persistência das dejeções e da colúria.
Ao longo do internamento e tendo em conta a clínica e alterações analíticas apresentadas consideraram-se várias hipóteses diagnósticas, nomeadamente PTT, SHUt, doença infeciosa (viagem transatlântica recente e residência em local com vários lagos e águas paradas), doença neoplásica com síndrome neoplásica associada (pelos antecedentes de neoplasia e idade) e SHUc.
Apresenta-se na Tabela 1 os exames complementares de diagnóstico realizados para estudo etiológico.

Dada a forte suspeita inicial de PTT, realizou uma sessão de permuta plasmática (com plasma fresco) por dia, perfazendo um total de seis sessões, com melhoria da função renal, mas persistência de anemia e trombocitopenia (Tabela 2). Fez 3 pulsos de 500 mg de metilprednisolona e posteriormente prednisolona 1 mg/kg/dia. A doente teve necessidade de suporte transfusional após três das seis sessões de permuta plasmática, por agravamento da anemia (Hb < 7 g/dL). Dada a evolução paulatina com a permuta plasmática, os resultados negativos de E. coli e toxina Shiga e a atividade normal da ADAMTS13, tornou-se premente considerar outros diagnósticos, nomeadamente a SHUc, tendo-se solicitado a autorização para administração de eculizumab.
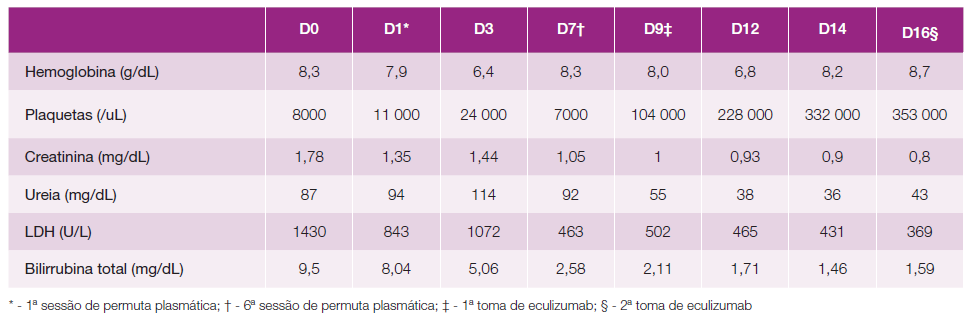
Ao nono dia de internamento (data em que foi disponibilizado o fármaco, após aprovação pela farmácia hospitalar) realiza primeira toma de eculizumab (900 mg), num esquema de quatro tomas programadas, com melhoria franca da anemia e trombocitopenia e recuperação da função renal (Tabela 2). Administrada vacina meningocócica e pneumocócica e iniciada profilaxia com ciprofloxacina 500 mg 1/dia durante duas semanas e cotrimoxazol 960 mg 3/semana, de acordo com recomendações.
Durante o internamento no SMI, a doente não desenvolveu falência respiratória ou cardiovascular, nem houve necessidade de suporte ventilatório ou hemodinâmico.
A doente foi posteriormente transferida para a enfermaria, mantendo boa evolução clínica, com consolidação da recuperação da função renal e resolução da trombocitopenia e anemia. À data da alta, foi conhecido o estudo genético do complemento que não identificou mutações nem polimorfismos de risco, mantendo-se, no entanto, a indicação de realização em hospital de dia da quarta toma de eculizumab.
Discussão
A SHUc, apesar de raro, deve ser um diagnóstico a considerar num adulto ou criança com um quadro de microangiopatia trombótica, com doseamento normal de ADAMTS13 e pesquisa de toxina Shiga negativa.
A clínica apresentada à admissão pela doente levou-nos à primeira hipótese diagnóstica de PTT, tendo a abordagem terapêutica inicial sido dirigida nesse sentido. Com a evolução clínica, analítica e com o surgimento dos resultados laboratoriais, as principais causas foram sendo descartadas (nomeadamente PTT e SHUt), levando os autores, após revisão exaustiva, a considerar a hipótese de SHUc.
O gold-standard do tratamento da SHUc é o eculizumab que deverá ser iniciado nas primeiras 24 a 48 horas da doença, o que nem sempre é possível devido à burocracia envolvida na aprovação deste fármaco.2,7No caso da doente apresentada entre a solicitação do fármaco à comissão de farmácia hospitalar, a aprovação pela administração hospitalar e a disponibilização do mesmo pela farmacêutica, passaram sete dias o que implicou a manutenção de sessões de permuta plasmática, como única forma terapêutica disponível.
Dada a gravidade clínica e analítica apresentada pelos doentes, na maioria dos casos impõe-se o início de tratamento com permuta plasmática, uma vez que se torna a solução terapêutica mais acessível e disponível no imediato (apenas com a única condicionante da transferência da doente para um centro hospitalar terciário). Ao contrário do eculizumab a resposta à permuta plasmática é habitualmente limitada.1,3,5 No caso descrito, e contrariamente ao expectável numa fase inicial houve uma melhoria clínica e analítica com esta técnica, no entanto com o início do eculizumab observamos uma melhoria significativa dos valores de hemoglobina, plaquetas, função renal e LDH, o que se encontra em concordância com a literatura, uma vez que esta prevê recuperação quase total.1,2,5,8
Relativamente ao estudo genético do complemento, neste caso não foram identificadas mutações ou polimorfis-mos, como é descrito em até 50% dos casos.4 Este achado levanta a questão da permanência ou suspensão do eculizumab.1,5,8Pela gravidade do quadro clínico e ausência de melhoria substancial, com o intuito de evitar a recidiva, mantiveram-se a quarta e quinta semanas de tratamento com 900 e 1200 mg de eculizumab respetivamente.
Com este caso os autores pretendem sensibilizar a comunidade clínica para a presença desta doença, que apesar de rara, apresenta elevada morbi-mortalidade e exige uma gestão clínica e terapêutica complexas. Na suspeita de PTT, pela gravidade do quadro clínico, a prioridade passará pelo início da terapêutica com permuta plasmática. A não resposta favorável fez questionar o diagnóstico e adequar a terapêutica com a utilização do eculizumab que, apesar de dever ser iniciada nas primeiras 48 horas tem um tempo de disponibilização mais prolongado. Para os doentes em que não é identificada qualquer mutação ou polimorfismo, são necessários mais estudos que apoiem a decisão terapêutica mais adequada, nomeadamente a duração, periodicidade ou suspensão do tratamento.

