










Servicios Personalizados
Revista
Articulo
 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkJornal Português de Gastrenterologia
versión impresa ISSN 0872-8178
J Port Gastrenterol. vol.20 no.5 Lisboa set. 2013
https://doi.org/10.1016/j.jpg.2013.03.003
CASO CLÍNICO
Doença de Wilson numa criança obesa
Wilson disease in an obese child
Mariana Domingues∗, Sandra Ferreira e Isabel Goncalves
Unidade de Transplante Hepático, Hospital Pediátrico Carmona da Mota, Centro Hospitalar e Universitário de Coimbra, Coimbra, Portugal
*Autor para correspondência
RESUMO
Apresenta-se o caso de uma criança assintomática referenciada por suspeita de fígado gordo não alcoólico associado a história prolongada de excesso de peso/obesidade. Apresentava elevação das enzimas de citólise hepática e ecografia abdominal compatível com esteatose do fígado. Apesar de perda ponderal inicial, acompanhada de descida das enzimas, a deteção de um valor de ceruloplasmina inferior ao limite do normal obrigou a investigação adicional, incluindo estudos de genética e biologia molecular, que confirmaram Doença de Wilson. Um terço das crianças obesas tem fígado gordo não alcoólico. Uma percentagem reduzida terá associada outra patologia.
A Doença de Wilson é um distúrbio hereditário do metabolismo do cobre com um largo espectro clínico. Nas crianças predominam as manifestações hepáticas, pelo que esta patologia deve ser sistematicamente excluída em doentes com alterações hepáticas não enquadráveis nos diagnósticos mais habituais. Sendo uma doença tratável, sublinha-se a necessidade de manter o elevado nível de suspeição para que se possa intervir precocemente.
Palavras-Chave: Doença de Wilson; Obesidade; Cobre; ATP7B; Idade pediátrica; Doença hepática
ABSTRACT
We present a case of a symptom-free girl with a long history of overweight/obesity, referred for suspected nonalcoholic steatohepatitis. She presented elevated liver enzymes and a liver ultrasound with steatosis. Despite initial weight loss and the improvement of liver enzymes, the detection of a low caeruloplasmine level lead to further evaluation, including genetic and molecular biology tests, which confirmed Wilson Disease. Most obese children have nonalcoholic steatohepatitis and a few will suffer from other disease.
Wilson Disease is an autossomal recessive disorder of copper metabolism with protean manifestation. Liver disease is the predominant presentation in children. Wilson Disease must be excluded in every cases of liver disease of unknown origin. Increased awareness of Wilson Disease in the pediatric population should lead to earlier diagnosis and improve the prognosis of this serious, but treatable, inborn error of metabolism.
Keywords: Wilson Disease; Obesity; Copper; ATP7B; Pediatric; Liver
Introdução
A Doença de Wilson (DW) é um distúrbio hereditário do metabolismo do cobre caracterizado pela disfunção da proteína responsável pelo transporte deste metal para a bílis (WDP). A acumulação progressiva no organismo, particularmente no fígado e cérebro1, induz lesão tóxica inflamatória e mitocondrial com espetro clínico muito variável. O gene mutado, ATP7B (cromossoma 13), é responsável pela síntese da proteína transportadora de cobre. Atualmente conhecem-se mais de 300 mutações patogénicas2,3. A incidência esta doença, de acordo com a sintomatologianeurológica, está estimada em 1/5.000 e 1/30.0002,4, com o gene mutado detetado com uma frequência de 1/90 a1/150 na população geral5. Na base de dados do EuroWilson (http://www.eurowilson.org/), com registos desde 2005, atribui-se a Portugal uma incidência média de 0,93/1.000.0006.
O diagnóstico de DW na população pediátrica reveste-se de alguma complexidade7. Muitas vezes, o atraso no diagnóstico fica a dever-se à variabilidade da apresentação clínica. Contudo, nesta idade, o envolvimento hepático prevalece como a principal forma de apresentação, de tal modo que o diagnóstico de DW deve ser sistematicamente excluído na doença hepática da criança, aguda ou crónica6.
Descreve-se o caso de uma criança de 10 anos obesa com fígado gordo não alcoólico (FGNA) em que se realizou o diagnóstico de DW em fase pré-sintomática. A associação entre obesidade e DW é mal conhecida. Ambas partilham esteatose hepática ou esteatohepatite, o que numa fase inicial da doença pode dificultar o diagnóstico diferencial.
Caso clínico
Criança de 10 anos, sexo feminino, primeira filha de pais não consanguíneos. Crescimento adequado (percentil 50 para peso e estatura) até aos 15 meses; progressivamente, excesso de peso e, a partir dos 4 anos, obesidade (IMC > P95). A estatura aproximou-se do P95 aos 7 anos de idade, onde ainda se mantém. Foram detetados vários erros alimentares, em quantidade e qualidade, e delineadas estratégias para perda de peso, sem sucesso. Dos antecedentes familiares, destaca-se história de várias gerações com excesso de peso e obesidade (tanto do lado materno como paterno) e doença do foro psiquiátrico, grave e mal controlada, numa tia materna.
No decurso do seu seguimento habitual, aos 10 anos de idade, foram detetados, na avaliação laboratorial valores elevados da aspartato aminotransferase (AST) e da alanina aminotransferase (ALT), 114 e 326 UI/L, respetivamente. A ecografia abdominal identificou «fígado globoso, de contornos regulares, com ecoestrutura homogénea mas acentuadamente hiperecogénica, com vesícula biliar normal, sem cálculos nem lama biliar e com vias biliares não ectasiadas». Não apresentava qualquer sintomatologia e não foram referidas alterações do comportamento da caligrafia e /ou rendimento escolar.
Foi então referenciada à consulta de hepatologia por obesidade, elevac¸ão das enzimas de citólise hepática e esteatose hepática em ecografia abdominal. O exame objetivo inicial, incluindo o exame neurológico sumário, era normal, excetuando-se a obesidade (peso -66 kg (> P 95), estatura - 154 cm (P 95) e Índice de Massa Corporal (IMC) - 28,6 kg/m2 (> P 95)).
Foi-lhe recomendado e ensinado um regime alimentar mais adequado e exercício físico. Reavaliada 2 meses depois, verificou-se perda ponderal de cerca de 1 kg acompanhada de uma descida de 60-70% no valor das aminotransferases. A investigação laboratorial para a esteatohepatite entretanto realizada revelou níveis plasmáticos de ceruloplasmina bastante baixos, de 0,023 g/L (N: 0,20 - 0,60 g/L), e cobre urinário basal de 48mg (N < 100mg/24 h). Não havia hemólise nem tubulopatia. Perante tais dados, efetuada prova com D-penicilamina, que foi positiva (464<< mg/24 h).
A hipótese de se tratar de uma DW foi posteriormente corroborada pela biopsia hepática, com valor de cobre de 606<< mg por grama de fígado seco (N < 50mg/g) e lesão do tipo hepatite crónica com esteatose e fibrose septal completa.
Observada pela oftalmologia, apresentava também esboço de anéis de Kayser-Fleischer bilateralmente. O exame neurológico detalhado em consulta de neuropediatria não revelou alterações.
A ressonância magnética nuclear cranioencefálica e a densitometria óssea realizadas foram normais (Z-score de −0,4).
A pesquisa de mutações em toda a região codificante do gene ATP7B por polimerase chain reaction (PCR) e por sequenciação direta (Genomed) identificou a mutação c.3402delC (p.Ala1135GInfsX13) em homozigotia no exão 15 e o polimorfismo c.2973G>A em homozigotia no mesmo gene.
Perante um score de Ferenci de 9 estabeleceu-se o diagnóstico de DW (http://www.eurowilson.org/professional/diagnosis/index.phtml#Scoring-system).
Iniciou terapêutica com acetato de zinco (que já suspendeu) e D-penicilamina (doses progressivas) e suplemento de piridoxina. Com um tempo de seguimento de 18 meses, mantém obesidade, cujo IMC é 25,8 kg/m2. Laboratorialmente, AST de 35 UI/L, ALT de 98 UI/L e cobre urinário de 42 ug/ 24 h. Mantém-se sem manifestações neurológicas da doença.
Discussão
O FGNA é atualmente a principal causa de doença hepática nas crianças e adolescentes no mundo industrializado, com uma importância crescente em paralelo com a «epidemia» de obesidade8. À medida que a sua prevalência cresce na idade pediátrica, impõe-se a necessidade de estandardização na abordagem clínica8. Perante a evidência laboratorial e ecográfica de esteatohepatite no contexto de obesidade da criança descrita, este seria o primeiro diagnóstico a considerar. De facto, esta suspeita deve existir em qualquer criança com mais de 3 anos de idade e excesso de peso/obesidade em que se verifique aumento do perímetro abdominal, sobretudo se houver história familiar positiva. A suspeita aumenta acima dos 10 anos8. A afirmação deste diagnóstico implica a exclusão de outras causas de hepatopatia. Do ponto de vista histológico, a DW faz parte do diagnóstico diferencial de esteatose ou esteatohepatite não alcoólica, sobretudo acima dos 10 anos de idade.
A DW, distúrbio do metabolismo do cobre de transmissão autossómica recessiva, foi descrita há um século por Kinnear Wilson9.
Segundo Harrison (1984), apesar de a DW se caracterizar por um largo espectro de apresentação clinica, o diagnóstico é acessível, desde que haja suspeita10. As manifestações hepáticas e neuropsiquiátricas são as mais comuns. A hepatopatia é mais frequente em crianças e jovens do que em adultos. No entanto, a sintomatologia é frequentemente inespecífica em qualquer idade9,11. O envolvimento hepático é variável, desde elevação assintomática das aminotransferases até apresentações com falência hepática aguda. O caso descrito enquadra-se no limite assintomático do espectro. O diagnóstico foi acidental e baseado em alterações laboratoriais na sequência de investigação de um quadro de obesidade. A elevação das aminotransferases e a esteatose hepática sugeriam um quadro de FGNA (ALT>AST). Um exame dirigido revelou anéis de Kayser-Fleischer.
Atualmente, o diagnóstico da DW faz-se com base na combinação de critérios clínicos, bioquímicos e genéticos12, segundo o sistema de pontuação proposto em 2001, na VIII Conferência Internacional de DW, o score de Ferenci. O doseamento de ceruloplasmina plasmática aceita-se como teste de rastreio na suspeita da doença11. No entanto, em idade pediátrica e/ou pacientes assintomáticos, só 50% dos doentes com DW têm ceruloplasmina baixa3,13.
Apesar dos recentes avanços no diagnóstico molecular, a biopsia hepática com obtenção de valores de cobre por grama de peso seco do fígado superiores a 250mg continua a ser o método gold standard12. O diagnóstico diferencial a considerar nos casos de cobre hepático elevado em idade pediátrica é o de hepatite autoimune (HAI). Embora 20% dos doentes com DW apresente doseamento de anticorpos antinucleares (ANA) com valor positivo, a inflamação da HAI é, em regra, mais grave e associada a colestase histológica. Para além disso, a aplicação do score de HAI14 auxilia no diagnóstico diferencial. Doenças mais raras, associadas a retenção de cobre (copper related liver disorders), cursam com ceruloplasmina normal ou elevada15.
Nos doentes sintomáticos, a excreção urinária de cobre das 24 h é tipicamente superior a 100mg9. Nos assintomáticos, essa excreção é menor. Sugere-se atualmente que a excreção urinária de cobre superior a 40mg/24 h (ainda que inferior a 100mg/24 h) é sugestiva de DW em crianças assintomáticas. Nestas, a prova com D-penicilamina não parece revelar qualquer papel diagnóstico7, ao contrário do que acontece nas sintomáticas. Esta poderá ser a explicação para a resposta à D-penicilamina no caso reportado, já que se trata de uma doente assintomática. Embora o cobre urinário tenha aumentado em 10 vezes após a prova, permaneceu inferior ao valor considerado diagnóstico. Histologicamente, as alterações são variadas e muitas vezes inespecíficas. As anomalias morfológicas mais precoces da DW são a presença de esteatose hepática micro e/ou macrovesicular, núcleos glicogenados nos hepatócitos periportais e necrose hepatocelular focal (fig. 1). Com a lesão e inflamação progressivas, instala-se fibrose e, progressivamente, cirrose (micronodular ou mista)5.
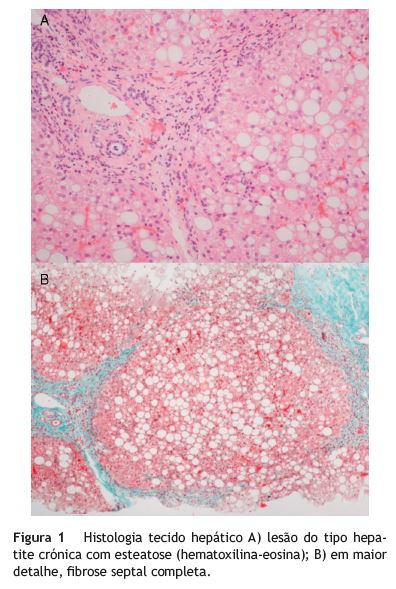
O valor diagnóstico dos anéis de Kayser Fleisher (KF) varia consoante a DW se manifesta com doença predominantemente hepática ou neurológica11,12. Num estudo efetuado por Steindl et al., apenas 50% dos doentes com hepatopatia tinham anéis de KF, contra 90% nos doentes com doença neurológica12. Na primeira década de vida raramente estão presentes. Os depósitos iniciais detetados na doente estão de acordo com a idade e evolução da doença.
Para além do diagnóstico, uma parte importante da DW é a monitorização da terapêutica (ceruloplasmina, cobre urinário das 24 h e cálculo do cobre livre no mínimo a cada 6 meses)9. O objetivo é, por um lado, avaliar a adesão ao tratamento e, por outro, evitar a depleção de cobre.
Numa criança com FGNA, se a perda de peso se acompanha de redução das enzimas é menos provável a DW. No caso descrito verificou-se redução de enzimas após perda ponderal, mas a deteção de ceruloplasmina baixa condicionou investigação adicional. A história familiar de doença psiquiátrica mal caracterizada foi também relevante para a investigação, embora se desconhecesse a etiologia (genética molecular para DW em curso).
Um estudo recente, tentando descrever a patogenia da FGNA, concluiu que o metabolismo do cobre não é relevante16. Portanto, se num doente obeso com FGNA se verificarem alterações do metabolismo do cobre, a hipótese de DW é a mais provável. Não se conhece uma relação preferencial entre obesidade, FGNA e DW. Em 2009, um grupo austríaco analisou essa relação, encontrando 2 casos de DW em 291 crianças e adolescentes obesos incluídos no estudo, o que representa uma maior incidência da doença do que o previamente estimado17. Provavelmente, a incidência de DW no universo crescente dos obesos será sobreponível à da população geral na mesma área geográfica. Em Portugal, a incidência de DW permanece desconhecida, estando seguramente subestimada no registo Eurowilson. O número de crianças e adolescentes obesos tem vindo a aumentar, pelo que é previsível que neste grupo de doentes se registe um número igualmente crescente de DW. Teremos de equacionar o custo-benefício do rastreio de DW nas crianças e adolescentes com FGNA e esteatohepatite.
Em conclusão, é importante sublinhar que, embora o FGNA seja a principal causa de hepatopatia em idade pediátrica e cada vez mais frequente, a afirmação do seu diagnóstico implica a exclusão de outras causas. Embora existam já linhas de orientação diagnóstica no FGNA da criança, a abordagem deve ser faseada e orientada de acordo com a faixa etária e os elementos clínicos recolhidos na história8. O índice de suspeição para DW deve ser elevado sempre que se verifique hepatopatia e/ou um distúrbio do sistema nervoso central de etiologia desconhecida, num indivíduo entre os 10-40 anos6. De facto, deve ser sistematicamente excluída em doentes com alterações hepáticas não enquadráveis nos diagnósticos mais habituais3. Obesidade e DW partilham a mesma lesão histológica, pelo que o doseamento de cobre hepático é um elemento importante para distinguir as duas entidades, quando a biopsia for necessária na investigação de um doente obeso. A DW tem disponível tratamento eficaz11. O diagnóstico tardio acompanha-se de alterações que podem ser irreversíveis culminando em transplante hepático.
Bibliografia
1. Pfeiffer R. Wilsons Disease. Semin Neurol. 2007;27:123-32. [ Links ]
2. Gupta A, Chattopadhyay I, Mukherjee S, Sengupta M, Das K, Ray SK. A novel COMMD1 mutation Thr174Met associated with elevated urinary copper and signs of enhanced apoptotic cell death in aWilson Disease patient. Behav Brain Funct. 2010;6:33. [ Links ]
3. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Wilsons Disease. J Hepatol. 2012;56:671-85. [ Links ]
4. Taylor R, Chen Y, Dhawan A. Triethylene tetramine dihydrochloride (trientine) in children with Wilson disease: Experience at Kings College Hospital and review of the literature. Eur J Pediatr. 2009;168:1061-8. [ Links ]
5. Ferenci P. Wilsons Disease. Clin Liver Dis. 1998;2:31-49. [ Links ]
6. Slovis TL, Dubois RS, Rodgerson DO, Silverman A. The varied manifestations of Wilsons Disease. J Pediatr. 1971;78:578-84. [ Links ]
7. Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Reevaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010;52:1948-56. [ Links ]
8. Vajro P, Lenta S, Socha P, Dhawan A, McKiernan P, Baumann U, et al. Diagnosis of NAFLD in children and adolescents. J Pediatr Gastroenterol Nutr. 2012;54:700-13. [ Links ]
9. Roberts E, Schilsky M, AASLD. Diagnosis and treatment of Wilson Disease: An update. Hepatology. 2008;47:2089-111. [ Links ]
10. Prado A, Fonseca D. Uma revisão sobre a doença de Wilson --relato de caso. Saúde. 2004;30:69-75. [ Links ]
11. Sánchez-Albisua I, Garde T, Hierro L, Camarena C, Frauca E, de la Vega A, et al. A high index of suspicion: The key to an early diagnosis of Wilsons disease in childhood. J Pediatr Gastroenterol Nutr. 1999;28:186-90. [ Links ]
12. EuroWilson (consultado 24 Set 2012). Disponível em: http://www.eurowilson.org/professional/diagnosis/index.phtml#Scoring-system [ Links ]
13. Dhawan A, Taylor RM, Cheeseman P, de Silva P, Katsiyiannakis L, Mieli-Vergani G. Wilsons disease in children: 37-year experience and revised Kings score for liver transplantation. Liver Transpl. 2005;11:441-8. [ Links ]
14. Manns MP, Czaja AJ, Gorham JD, Krawitt EL, Mieli-Vergani G, Vegani D, et al. Diagnosis and management of Autoimmune Hepatitis. Hepatology. 2010;51:2-9. [ Links ]
15. Muller T, Muller W, Feichtinger H. Idiopathic copper toxicosis. Am J Clin Nutr. 1998;67 Suppl:1082-6. [ Links ]
16. Akin K, Beyler AR, Kaya M, Erden E. The importance of iron and copper accumulation in the pathogenesis of non-alcoholic steatohepatitis. Turk J Gastroenterol. 2003;14:228-33. [ Links ]
17. SpindelboeckW, Deutschmann A, Lackner K,Weitzer C, Erwa W, Hauer A, et al. Obesity and Wilson disease in childhood and adolescence. J Pediatr Gastroenterol Nutr. 2009;48 Suppl 3:E63. [ Links ]
Responsabilidades éticas
Proteção dos seres humanos e animais. Os autores declaram que os procedimentos seguidos estavam de acordo com os regulamentos estabelecidos pelos responsáveis da Comissão de Investigação Clínica e Ética e de acordo com os da Associação Médica Mundial e da Declaração de Helsinki.
Confidencialidade dos dados. Os autores declaram ter seguido os protocolos do seu centro de trabalho acerca da publicação dos dados de pacientes e que todos os pacientes incluídos no estudo receberam informações suficientes e deram o seu consentimento informado por escrito para participar nesse estudo.
Direito à privacidade e consentimento escrito. Os autores declaram ter recebido consentimento escrito dos pacientes e/ ou sujeitos mencionados no artigo. O autor para correspondência deve estar na posse deste documento.
Conflito de interesses
Os autores declaram não haver conflito de interesses.
*Autor para correspondência
Correio eletrónico: mariana.dominguess@gmail.com (M. Domingues).
Recebido a 14 de novembro de 2012; aceite a 7 de março de 2013

{kind=link}