










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Pneumologia
versão impressa ISSN 0873-2159
Rev Port Pneumol v.16 n.5 Lisboa set. 2010
Discinesia ciliar primária revisitada: A propósito de três casos clínicos
Joana Fermeiro 1, Teresa Bandeira 2, Luísa Lobo 3 e Luísa Pereira 2
1 Interna do Internato Complementar de Pediatria
2 Assistente Hospitalar Graduada de Pediatria
3 Assistente Hospitalar Graduada de Radiologia
Unidade de Pneumologia Pediátrica, Cuidados Respiratórios Domiciliários e de Transição do HSM
Departamento da Criança e da Família do Hospital de Santa Maria – Centro Hospitalar Lisboa Norte, EPE
Director: Professor Doutor Paulo Magalhães Ramalho
Resumo
A discinesia ciliar primária é uma doença genética e clinicamente heterogénea, cuja patogénese assenta em compromisso da estrutura e função ciliares.
Cursa habitualmente com infecções respiratórias recorrentes, infertilidade e ainda situs inversus total em cerca de metade dos doentes.
Dada a raridade e heterogeneidade fenotípica desta patologia o seu diagnóstico implica frequentemente um elevado índice de suspeição.
Com este artigo os autores procuraram rever os aspectos fundamentais relativos à etiopatogénese, quadro clínico e abordagens diagnóstica e terapêutica da discinesia ciliar primária, contextualizando-os na discussão de três casos clínicos.
Descrevem-se três doentes cujo quadro clínico inaugural foi marcado por síndroma de dificuldade respiratória neonatal associada, em dois doentes, a situs inversus total. A evolução clínica subsequente cursou com sintomatologia respiratória em dois doentes (tosse produtiva crónica num doente e episódios recorrentes de pneumonia e sibilância noutro) e atingimento do aparelho respiratório superior nos três. À data do diagnóstico de discinesia ciliar primária, os doentes apresentavam idades distintas (8 meses, 5 e 12 anos), sendo que os dois doentes com idades de diagnóstico mais tardias apresentavam nessa altura compromisso funcional respiratório do tipo obstrutivo.
Os autores discutem os diferentes padrões de apresentação clínica, a abordagem terapêutica e a evolução clínica subsequente, os factores potencialmente implicados no diagnóstico tardio e as suas repercussões prognósticas.
O objectivo primordial é alertar para manifestações clínicas precoces e/ou frequentes de discinesia ciliar primária, procurando deste modo influenciar o prognóstico pela melhoria do reconhecimento precoce desta patologia.
Palavras-chave: Discinesia ciliar primária, situs inversus total.
Primary ciliary dyskinesia revisited: Based on three clinical reports
Abstract
Primary ciliary dyskinesia is a genetically and clinically heterogeneous disorder. Its pathogenesis reflects structural and functional compromise of the cilia.
Common clinical manifestations include recurrent upper and lower respiratory tract infections and infertility, as well as situs inversus totalis in half of the affected patients.
Besides its rarity and phenotypic heterogeneity its diagnosis usually requires a high suspicion index.
The main purpose of this paper is to review the pathogenesis, clinical features, diagnostic and therapeutic approaches of primary ciliary dyskinesia beyond the discussion of three clinical reports.
We report the cases of three patients all with a past history of neonatal respiratory distress and two with situs inversus totalis. The subsequent clinical manifestations included lower airway symptoms in two patients (chronic productive cough and recurrent pneumonia and wheezing) and upper respiratory tract disease in all patients. Age at primary ciliary dyskinesia diagnosis differed considerably among patients (8 months, 5 and 12 years). The two patients with later diagnosis had already obstructive lung function compromise at the time of diagnosis.
The authors discuss the different clinical patterns presented, therapeutic strategies and the clinical progression that ensued, factors possibly implicated in late diagnosis and its prognostic consequences.
The main goal is to emphasize early and/or prevalent clinical features of primary ciliary dyskinesia in order to promote clinical awareness and early recognition of the disease.
Key-words: Primary ciliary dyskinesia, situs inversus totalis.
Introdução
A discinesia ciliar primária (DCP) é uma doença genética rara, associada predominantemente a um padrão de hereditariedade autossómico recessivo, embora tenha sido documentada, em alguns casos, transmissão ligada ao cromossoma X1-3.
A apresentação clínica é heterogénea e a etiopatogenia caracterizada por alterações estruturais e/ou funcionais ciliares que condicionam disfunção da mobilidade dos cílios das células epiteliais respiratórias, cílios ganglionares embrionários e flagelos dos espermatozóides4,5.
Estima-se em 1:15 000-30 000 nados-vivos a prevalência de DCP, embora se considere que a prevalência real possa ser superior, atribuível ao subdiagnóstico6,7.
A DCP cursa com um espectro fenotípico alargado e heterogéneo que inclui combinações variáveis de manifestações clinicoima giológicas1,8 com atingimento multiórgão e expressividade individual variável (Quadro I).
Quadro I – Manifestações clínicas de discinesia ciliar primária

A repercussão da discinesia ciliar primária a nível pulmonar assume particular relevância prognóstica9. A ineficácia da clearance mucociliar determina infecções respiratórias recorrentes associadas a processo inflamatório crónico predominantemente neutrofílico com progressão habitual para lesão pulmonar irreversível com bronquiectasias, culminando nalguns casos em insuficiência respiratória crónica4,10.
A apresentação clínica pode ser inespecífica, sobreponível à clínica de bronquiectasias, o que frequentemente leva à necessidade de diagnóstico diferencial com fibrose quística. A associação com envolvimento de outros órgãos ou sistemas, nomeadamente situs inversus total ou atingimento do aparelho respiratório superior, com ocorrência de otite média serosa persistente, hipoacusia de transmissão (particularmente prevalente em idade pediátrica) e sinusite conduzem de forma mais fácil à suspeição diagnóstica7,11.
Quando ocorre simultaneamente, a tríade de situs inversus total, bronquiectasias e sinusite, esta é conhecida como síndroma de Kartagener1,12.
Outros órgãos atingidos, por disfunção dos cílios não móveis em doentes com DCP, são o rim, o fígado e o olho, com aparecimento de doença poliquística renal/hepática, atresia biliar ou patologia da retina11,13. As manifestações clínicas e imagiológicas incaracterísticas determinam a necessidade de estabelecimento de diagnóstico definitivo através de microscopia electrónica de transmissão ou videomicroscopia de alta velocidade para confirmação, respectivamente, de alteração ultraestrutural ciliar e/ou compromisso funcional ciliar1,7.
Apresentam-se três casos clínicos que, pela diversidade da apresentação clínica, da idade no diagnóstico e da extensão das lesões, poderão contribuir para a melhoria dos critérios de suspeição e orientação nesta doença rara.
Casos clínicos
Caso clínico 1
Adolescente do sexo masculino, actualmente com 12 anos, referenciado à consulta de Pneumologia Pediátrica aos 5 anos por hipótese de DCP.
Gestação de termo, sem consanguinidade parental ou outros antecedentes familiares de relevo. Foi internado após o nascimento numa unidade neonatal por síndroma de dificuldade respiratória nas primeiras horas de vida, com necessidade de instituição de oxigenoterapia suplementar. Manteve necessidade de oxigenoterapia suplementar no domicílio até aos 1,5 meses de idade. O ecocardiograma realizado nesta altura documentou dextrocardia, sem cardiopatia estrutural associada. A ecografia abdominal confirmou o diagnóstico de situs inversus total.
Aos 3 meses teve novo internamento hospitalar de curta duração por bronquiolite aguda. Posteriormente, verificou-se início de episódios recorrentes de tosse produtiva. Por diminuição da acuidade auditiva iniciou seguimento regular em consulta de Otorrinolaringologia aos 3,5 anos, tendo sido diagnosticada hipoacusia de transmissão no contexto de otite média serosa bilateral.
Apresentava bom estado geral e parâmetros antropométricos adequados (peso P75 -90 e estatura P90), saturação percutânea de oxigénio (SpO2) de 94% e ausência de dispneia ou sinais de dificuldade respiratória, deformação torácica ou hipocratismo digital. À auscultação pulmonar verificava-se existência de murmúrio vesicular simétrico sem ruídos adventícios.
Foi realizado estudo imunoalergológico, tendo os doseamentos de imunoglobulina (Ig) E total, IgE específicas para alergénios inalantes, IgA, IgM e IgG (incluindo subclasses) sido normais.
O estudo ultraestutural de cílios do epitélio efectuou-se com ecovagens da mucosa nasal ao nível do corneto inferior, tendo o exame ultraestrutural dos cílios revelado ausência de braços externos e internos de dineína, confirmando definitivamente o diagnóstico de discinesia ciliar primária.
Verificou-se evolução clínica subsequente favorável, destacando-se ocorrência de apenas três episódios de exacerbação respiratória, com boa resposta à antibioticoterapia oral instituída em ambulatório e progressão estatural e ponderal nos percentis 90 e 75-90, respectivamente, e índice de massa corporal (IMC) P50-75.
O estudo funcional respiratório efectuado aos 6 anos revelou padrão de insuflação e de obstrução brônquica e bronquiolar com ausência de reversibilidade na prova de broncodilatação, que se mantiveram, tendo actualmente capacidade vital forçada (CVF) 2,62 L (79,9%), volume expiratório máximo por segundo (VEMS) 1,82 L (66,7%), débito expiratório máximo entre 25% e 75% da CVF (FEF 25-75) 1,19 L/s (36,9%) e volume residual (VR) 1,55 L (159%).
A tomografia computorizada de alta resolução (TC-AR) torácica, realizada aos 6 anos, documentou espessamento da parede dos brônquios centrais e atelectasia subsegmentar anterior na base esquerda. Relativamente à evolução sob o ponto de vista imagiológico, a TC-AR torácica efectuada aos 12 anos evidenciava insuflação pulmonar global, bronquiectasias cilíndricas com distribuição central e bilateral com espessamento da parede brônquica associado, atelectasia parcial do andar médio do pulmão esquerdo, com bronquiectasias no interior e alguns nódulos centrilobulares no lobo inferior do pulmão direito, em provável relação com infiltrado inflamatório e impactações bronquiolares (Fig. 1).
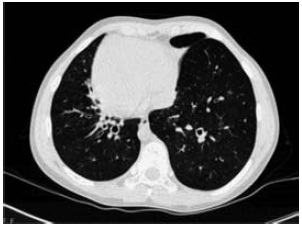
Fig. 1 – Bronquiectasias cilíndricas com distribuição central (tomografia computorizada de alta resolução torácica).
Caso clínico 2
Criança do sexo feminino, actualmente com 3 anos, primeira filha de pais não consanguíneos, sem antecedentes familiares relevantes. Gravidez medicamente assistida (inseminação artificial) por infertilidade materna primária, tendo o período gestacional decorrido sem intercorrências.
Parto eutócico às 37 semanas no Hospital de Santa Maria, sem necessidade de reanimação. Ao terceiro dia de vida verificou-se início de rinorreia anterior mucosa abundante, a que se associou, no dia seguinte, quadro de tosse e tiragem infracostal ligeira. Nessa altura realizou radiografia de tórax que evidenciou dextrocardia e avaliação analítica, cujos resultados não eram sugestivos de infecção bacteriana.
Verificou-se agravamento progressivo do quadro de dificuldade respiratória, com necessidade de aporte suplementar de oxigénio na incubadora (FiO2 22-27%) entre o 5.º e o 9.º dias de vida. Manteve ainda rinorreia anterior abundante, salientando-se aquisição gradual de aspecto purulento e necessidade de aspiração frequente de secreções nasais até à altura da alta (1 mês).
A prova de suor realizada por método semiquantitativo foi normal.
Efectuou também ecocardiograma e ecografia abdominal, que confirmaram situs inversus total.
Por hipótese diagnóstica de DCP iniciou acompanhamento na Unidade de Pneumologia Pediátrica do HSM aos 1,5 meses.
Nessa altura apresentava progressão estaturoponderal adequada (peso P50 e estatura P3-P10), ausência de sinais de dificuldade respiratória ou alterações relevantes à auscultação pulmonar, destacando-se ainda melhoria importante do quadro de rinorreia mucopurulenta.
O exame ultraestrutural dos cílios (amostra obtida mediante escovagens da mucosa nasal ao nível do corneto inferior) revelou ausência de braços externos e internos de dineína, tendo confirmado o diagnóstico de discinesia ciliar primária (Fig. 2).
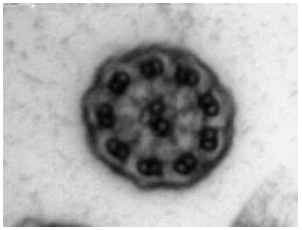
Fig. 2 – Ausência de braços internos e externos de dineína (microscopia electrónica).
A evolução clínica subsequente foi favorável, marcada por progressão estaturoponderal adequada e ausência de episódios de sibilância, tosse produtiva ou pneumonia até à data.
A radiografia de tórax realizada aos 13 meses revelou reforço do interstício peri-hilar bilateralmente.
Salienta-se ocorrência de episódio único de otite média aguda aos 2 anos e diagnóstico de otite média serosa bilateral aos 2,5 anos, no contexto de seguimento em consulta de Otorrinolaringologia previamente iniciado.
Caso clínico 3
Adolescente do sexo masculino, actualmente com 16 anos, sem antecedentes de consanguinidade parental ou outros antecedentes familiares relevantes. Gestação de termo, vigiada, sem intercorrências ou factores de risco infeccioso no período perinatal. Ao segundo dia de vida verificou-se início de quadro de dificuldade respiratória com necessidade de oxigenoterapia suplementar e broncorreia, no contexto de evidência radiológica de pneumonia. Por persistência de broncorreia foram realizados, ainda no período neonatal, prova de suor, pHmetria e doseamento de imunoglobulinas séricas, cujos resultados não apresentaram alterações relevantes.
Foi também realizado ecocardiograma que não evidenciou alterações estruturais e/ou funcionais.
A evolução clínica posterior cursou com episódios recorrentes de pneumonia e sibilância e sintomatologia sugestiva de sinusite. Aos 6 anos foi realizado estudo alergológico (negativo) e TC torácica, descrita como normal. Repetiu prova de suor, que foi normal, e espirometria que revelou padrão obstrutivo com resposta parcial na prova de broncodilatação. Face a persistência de episódios frequentes de pneumonia e sibilância recorrente sem melhoria após diferentes estratégias terapêuticas, foi referenciado à consulta de Pneumologia Pediátrica do HSM aos 12 anos.
À observação destacava-se bom estado geral, parâmetros antropométricos adequados (Peso P25 -50 e estatura P50-75), ausência de sinais de dificuldade respiratória ou hipocratismo digital. À auscultação pulmonar objectivaram-se sibilos inspiratórios e expiratórios e fervores crepitantes mobilizáveis com a tosse dispersos bilateralmente.
Foi realizada avaliação imunoalergológica (doseamentos de IgE total, IgE específicas para alergénios inalantes, IgG, IgM e IgA), que não apresentou alterações relevantes.
O estudo da ultraestrutura ciliar (cílios da mucosa nasal) por microscopia electrónica evidenciou ausência dos braços internos de dineína e irregularidades frequentes na configuração dos pares de microtúbulos, diagnósticas de DCP.
Verificou-se diminuição da frequência de episódios de agudização respiratória broncospástica, apresentando actualmente um a dois episódios anuais, não se tendo verificado necessidade de internamento.
O estudo funcional respiratório efectuado aos 12 anos revelou padrão de insuflação e de obstrução predominantemente bronquiolar com ausência de reversibilidade na prova de broncodilatação. Nos anos subsequentes manteve o padrão descrito, tendo actualmente CVF 4,53 L (107,5%t), VEMS 2,79 L (80,1%t), FEF 25-75 1,26 L/s (31,9%t) e VR 2,6 L (226,1%t).
A TC torácica realizada aos 12 anos documentou bronquiectasias na vertente inferior e interna da língula.
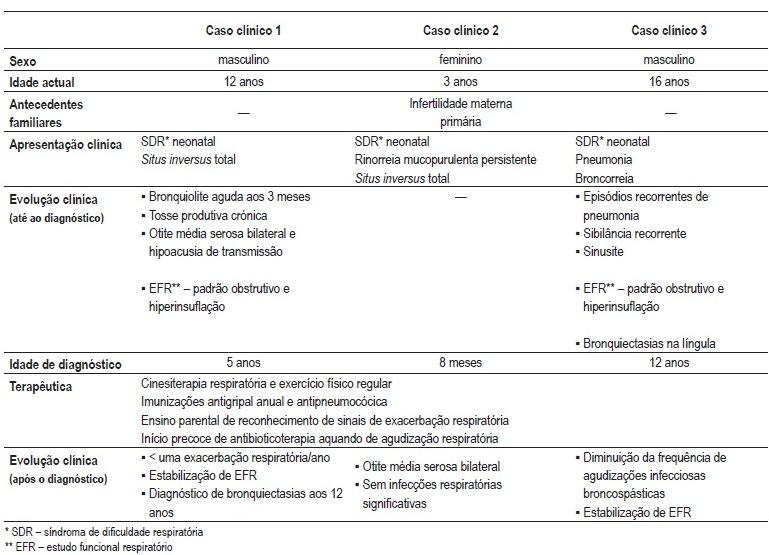
As estratégias preventivas e terapêuticas instituídas incluíram para todos os doentes: programa de reeducação funcional respiratória (manobras de percussão e vibração torácica nos casos clínicos 1 e 2 e técnica de vibração torácica interna associada a pressão expiratória positiva no caso clínico 3); incentivo da prática regular de exercício físico; imunizações antigipal anual e antipneu mocócica e início precoce de antibioticoterapia no contexto de agudização respiratória (Quadro II).
Quadro II – Quadro-resumo dos casos clínicos
Discussão
Os casos clínicos apresentados relatam doentes cujo quadro clínico teve início no período neonatal precoce. A síndroma de dificuldade respiratória, com necessidade de oxigenoterapia suplementar em recém-nascidos sem factor de risco identificável, constituiu a manifestação clínica inaugural nestes doentes.
No caso clínico 2, o quadro de dificuldade respiratória associou-se a rinorreia mucopurulenta contínua, que constitui uma manifestação característica de DCP no grupo etário em questão1,7.
Relativamente ao caso clínico 3, a síndroma de dificuldade respiratória neonatal enquadrava-se no diagnóstico de pneumonia sem factores de risco infeccioso identificáveis. Este é também considerado um padrão clínico evocativo de DCP1,7.
A síndroma de dificuldade respiratória neonatal não é classicamente englobada na descrição do fenótipo clínico característico da discinesia ciliar primária. Todavia, vários estudos de carácter retrospectivo documentam que uma proporção significativa dos doentes com esta patologia apresenta história pregressa de dificuldade respiratória neonatal (67% numa das séries), pelo que esta é actualmente reconhecida como forma de apresentação clínica frequente14.
Em dois dos casos clínicos descritos, foi ainda diagnosticado, no período neonatal, situs inversus total, sem cardiopatia estrutural associada.
No caso clínico 1, apesar da constelação de sintomas inaugurais sugestiva associada a situs inversus total, este diagnóstico foi equacionado tardiamente.
Já no caso clínico 2, um quadro clinicoimagio lógico inaugural análogo, distinguindo-se apenas pela associação de rinorreia anterior contínua, foi valorizado precocemente como sendo sugestivo de discinesia ciliar primária.
Os autores especulam que o facto de este segundo caso ter sido abordado, desde o início, no contexto de uma unidade terciária, tenha contribuído de forma decisiva para o diagnóstico precoce.
Num estudo retrospectivo realizado por Coren et al (publicado em 2002) foi avaliada a idade de diagnóstico e sintomatologia que antecedeu o diagnóstico em 55 crianças com discinesia ciliar primária11. Constatou-se que o diagnóstico de discinesia ciliar primária foi estabelecido antes dos 12 meses de idade em apenas 13 das 25 crianças com quadro de síndroma de dificuldade respiratória neonatal e situs inversus11.
Os autores desse artigo consideram que o diagnóstico tão frequentemente tardio de discinesia ciliar primária, mesmo no contexto de quadro clínico sugestivo de início precoce, assenta na raridade desta patologia e na ausência de conhecimento da mesma, agravada pela complexidade da sua confirmação diagnóstica11.
O facto de numa proporção significativa de doentes com discinesia ciliar primária o diagnóstico ser apenas estabelecido na idade adulta implica que o desafio diagnóstico não cessa na idade pediátrica e que é fundamental manter um índice de suspeição diagnóstica elevado aquando da observação de doentes adultos.
No doente do caso descrito 3, o fenótipo clínico, também fortemente sugestivo de discinesia ciliar primária era dominado por sintomatologia do foro respiratório, designadamente episódios recorrentes de pneumonia e de dificuldade respiratória. Este caso distinguia-se dos restantes devido à inexistência de situs inversus total concomitante.
Os autores consideram provável que esse facto tenha contribuído decisivamente para o diagnóstico tardio de discinesia ciliar primária constatado neste caso.
De facto, apesar de apenas 50% dos doentes com DCP apresentarem situs inversus total, a sua ausência eleva significativamente a dificuldade diagnóstica, uma vez que é reconhecido como um dos achados mais característicos desta patologia11.
Dada a raridade e heterogeneidade fenotípica da DCP, é fundamental que esteja sempre patente um elevado índice de suspeição diagnóstica4,7,15.
Face à inespecificidade da sintomatologia respiratória e à sua sobreposição a outras doenças com atingimento pulmonar crónico (essencialmente fibrose quística e imunodeficiência), a investigação de DCP frequentemente só é iniciada após exclusão destes diagnósticos, tal como sucedeu nos casos clínicos apresentados.
A determinação da ultraestrutura ciliar (epitélio nasal ou brônquico) por microscopia electrónica de transmisão constitui actualmente o método diagnóstico gold standard de DCP1.
Foram identificados diferentes defeitos ultraestruturais, sendo os mais frequentes a ausência dos braços externos de dineína, dos braços internos de dineína ou de ambos4,16.
Numa proporção de cerca de 15% de doentes com quadro imagiológico sugestivo de DCP, não se encontra evidência de alteração na ultraestrutura ciliar4,16. Nesses casos assume particular importância a identificação de disfunção ciliar, também diagnóstica de DCP, através do estudo da frequência e padrão de batimento ciliar, mediante videomicroscopia de alta velocidade1.
Noutras situações, o diagnóstico definitivo de DCP pode ser dificultado pela ocorrência concomitante de infecção ou inflamação respiratórias, que por constituírem causas secundárias e transitórias de disfunção ciliar poderão resultar em resultados falsos positivos1,17. Para minimizar a obtenção de resultados falsos positivos, é fundamental que a determinação da ultraestrutura e/ou função ciliares só seja realizada no contexto de quadro clínico fortemente sugestivo de DCP e sempre na ausência de infecção respiratória evidente7. Em caso equívocos, poder-se-á ainda recorrer à realização de ciliogénese em cultura celular, que possibilita a eliminação de eventuais lesões secundárias1.
Numa perspectiva histórica, o teste da sacarina constituiu durante décadas o método mais popular de rastreio de DCP1,17. O seu uso é bastante limitado em pediatria, uma vez que implica uma elevada capacidade de colaboração, nomeadamente a capacidade de permanecer sentado durante pelo menos uma hora7. Por outro lado, é um método pouco específico e que não permite a diferenciação entre disfunção ciliar primária ou secundária, não lhe sendo actualmente reconhecida utilidade diagnóstica8,14.
A medição do óxido nítrico (NO) nasal assume-se actualmente como a ferramenta de rastreio mais sensível e reprodutível, tendo ainda as vantagens adicionais de ser um método pouco invasivo e passível de ser realizado em crianças pequenas4,18. De facto, foram já realizados estudos em crianças com idade superior a 5 anos, que revelaram boa colaboração e reprodutibilidade de resultados elevada4,8. Nos doentes com DCP a produção de NO nasal encontra-se marcadamente diminuída. Considera-se que este achado aparentemente paradoxal (face à presença invariável de inflamação crónica na DCP) possa estar relacionado com a própria alteração da actividade ciliar ou expressão de isoformas de NO sintase alteradas4,8. A medição do NO nasal, significativamente diminuído nestes doentes, é então perspectivada como uma estratégia de rastreio promissora4,18.
Todavia, existe alguma sobreposição entre os valores de NO nasal encontrados em doentes com fibrose quística e DCP, pelo que o diagnóstico definitivo desta última requer sempre a realização de um dos métodos de diagnóstico descritos8.
A literatura actual é escassa em ensaios clínicos que avaliem a eficácia da maioria dos fármacos com benefício terapêutico potencial na discinesia ciliar primária15,19. De facto, a abordagem terapêutica da discinesia ciliar primária tem sido largamente extrapolada a partir da terapêutica de outras patologias que cursam com o aparecimento de bronquiectasias, nomeadamente da fibrose quística1,20.
Não existe tratamento curativo que permita corrigir ou reduzir a disfunção ciliar17,20, pelo que o objectivo primordial da terapêutica é adiar e, se possível, evitar o desenvolvimento de bronquiectasias e o declínio progressivo da função pulmonar9,10.
As estratégias terapêuticas habitualmente preconizadas centram-se, por um lado, em potenciar a clearanace mucociliar mediante o recurso a técnicas de cinesiterapia respiratória adaptadas individualmente e a promoção da prática regular de exercício físico e, por outro lado, no tratamento precoce e eficaz de exacerbações respiratórias1,7,15,17.
Outro pilar fundamental da abordagem terapêutica destes doentes é a prevenção de exacerbações respiratórias, mediante a imunização com vacinas antigripal e antipneumocócica1,15 e a evicção da exposição a agentes patogénicos respiratórios, tabagismo e a outros agentes irritantes inalantes que reconhecidamente se associem ao aumento de produção de muco15,17.
A abordagem terapêutica dos doentes apresentados neste artigo englobou as várias estratégias terapêuticas descritas.
Vários estudos sugerem que o início do compromisso da função respiratória em doentes com discinesia ciliar primária tem um início precoce na idade pediátrica9,21, pelo que a progressão da doença não tratada cursa com um declínio progressivo e significativo da função respiratória, que poderá culminar com instalação de insuficiência respiratória crónica4,10.
Na altura do diagnóstico do 1.º e 3.º doentes (aos 5 e 12 anos, respectivamente), estes apresentavam já compromisso funcional respiratório com padrão obstrutivo e insuflação.
Existe evidência crescente de que a implementação precoce de estratégias terapêuticas em centros especializados permite estabilizar a função pulmonar em doentes com bronquiectasias não relacionadas com FQ22, e nos doentes com DCP em particular9,23.
De facto, verificou-se estabilização da função pulmonar após a instituição de estratégias terapêuticas adequadas nestes dois doentes. A evolução constatada é sobreponível à de outras doenças respiratórias crónicas que cursam com o aparecimento de bronquiectasias não relacionadas com FQ22.
No caso clínico 2, diagnosticado antes do primeiro ano de vida, é expectável que a função pulmonar não sofra um declínio tão precoce e acentuado como constatado nos outros doentes, face ao início precoce de cinesiterapia respiratória e restantes atitudes preventivas e terapêuticas descritas.
Mesmo não existindo terapêutica curativa disponível, o estabelecimento precoce do diagnóstico de DCP repercute-se favoravelmente no seu prognóstico9,23.
São múltiplos os desafios inerentes ao diagnóstico e seguimento ulterior destes doentes, desafios esses que devem ser sempre abordados numa perspectiva multidisciplinar (pneumologia, otorrinolaringologia, fisiatria, genética e, noutra faixa etária, consulta de infertilidade), aproveitando a experiência e estrutura de centros especializados de fibrose quística.
Bibliografia
1. Bush A, Chodhari R, Collins N, Copeland F, Hall P, et al. Primary ciliary dyskinesia: current state of the art. Arch Dis Child 2007;92:1136-1140. [ Links ]
2. Narayan D, Krishnan SN, Upender M, Ravikumar TS, Mahoney MJ, et al. Unusual inheritance of primary ciliary dyskinesia (Kartageners syndrome). J Med Genet 1994; 31:493-496. [ Links ]
3. Iannaccone A, Breuer DK, Wang XF, Kuo SF, Normando EM, et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J Med Genet 2003; 40:e118. [ Links ]
4. Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, et al. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am J Respir Crit Care Med 2004; 169:459-467. [ Links ]
5. Loges NT, Olbrich H, Fenske L, Mussaffi H, Horvath J, et al. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am J Hum Genet 2008; 83:547-458. [ Links ]
6. Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol Pathol 2007; 35:116-129. [ Links ]
7. Bush A, Cole P, Hariri M, Mackay I, Phillips G, et al. Primary ciliary dyskinesia: diagnosis and standards of care. Eur Respir J 1998; 12:982-988. [ Links ]
8. Hogg C. Primary ciliary dyskinesia: when to suspect the diagnosis and how to confirm it. Paediatr Respir Ver 2009; 10:44-50. [ Links ]
9. Ellerman A, Bisgaard H. Longitudinal study of lung function in a cohort of primary ciliary dyskinesia. Eur Respir J 1997; 10:2376-2379. [ Links ]
10. Storm vans Gravesande K, Omran H. Primary ciliary dyskinesia: clinical presentation, diagnosis and genetics. Ann Med 2005; 37:439-449. [ Links ]
11. Coren ME, Meeks M, Morrison I, Buchdahl RM, Bush A. Primary ciliary dyskinesia: age at diagnosis and symptom history. Acta Paediatr 2002; 91:667-669. [ Links ]
12. Kartagener M, Stucki P. Bronchiectasis with situs inversus. Arch Pediatr 1962; 79:193-207. [ Links ]
13. Ong AC, Wheatley DN. Polycystic kidney disease: the ciliary connection. Lancet 2003; 361:774-776. [ Links ]
14. Ferkol T, Leigh M. Primary ciliary dyskinesia and newborn respiratory distress. Semin Perinatol 2006; 30:335-340. [ Links ]
15. Fauroux B, Tamalet A, Clément A. Management of primary ciliary dyskinesia: the lower airways. Paediatr Respir Rev 2009; 10:55-57. [ Links ]
16. Carda C, Armengot M, Escribano A, Peydró A. Ultrastructural patterns of primary ciliary dyskinesia syndrome. Ultrastruct Pathol 2005; 29:3-8. [ Links ]
17. Morillas HN, Zariwala M, Knowles MR. Genetic causes of bronchiectasis: primary ciliary dyskinesia. Respiration 2007; 74:252-263. [ Links ]
18. Lundberg JO, Weitzberg E, Nordvall SL, Kuylenstierna R, Lundberg JM, Alving K. Primarily nasal origin of exhaled nitric oxide and absence in Kartagener´s syndrome. Eur Respir J 1994; 7:1501-1504. [ Links ]
19. Lie H, Ferkol T. Primary ciliary dyskinesia: recent advances in pathogenesis, diagnosis and treatment. Drugs 2007; 67:1883-1892. [ Links ]
20. Amirav I, Cohen-Cymberknoh M, Shoseyov D, Kerem E. Primary ciliary dyskinesia: prospects for new therapies, building on the experience in cystic fibrosis. Paediatr Respir Rev 2009; 10:58-62. [ Links ]
21. Brown DE, Pittman JE, Leigh MW, Fordham L, Davis SD. Early lung disease in young children with primary ciliary dyskinesia. Pediatr Pulmonol 2008; 43:514-516. [ Links ]
22. Bastardo CM, Sonnappa S, Stanojevic S, Navarro A, Lopez PM, et al. Non-cystic fibrosis bronchiectasis in childhood: longitudinal growth and lung function. Thorax 2009; 64:246-251. [ Links ]
23. Leigh MW. Primary ciliary dyskinesia. Semin Respir Crit Care Med 2003; 24:653-662. [ Links ]
Departamento da Criança e da Família do Hospital de Santa Maria
Avenida Professor Egas Moniz
1649-035 Lisboa
e-mail: mluisafpereira@gmail.com
Recebido para publicação: 24.11.09
Aceite para publicação: 13.05.10
