










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Ortopedia e Traumatologia
versão impressa ISSN 1646-2122
Rev. Port. Ortop. Traum. vol.20 no.4 Lisboa dez. 2012
CASO CLÍNICO
Schwannomatose em mulher de 83 anos de idade
António MurinelloI; José SimõesII; Adelaide MilheiroIII; Ana MaçãsIV; Helena DamásioIV; Vasco RamalhoIV; António FigueiredoIV; Pedro GuedesIV; Ana SerranoIV
I. Serviço de Radiologia.Hospital Curry Cabral. Lisboa. Portugal.
II. Serviço de Anatomia Patológica. Hospital Curry Cabral. Lisboa. Portugal.
III. Serviço de Ortopedia. Hospital Curry Cabral. Lisboa. Portugal.
IV. Serviço de Medicina Interna. Hospital Curry Cabral. Lisboa. Portugal.
RESUMO
A schwannomatose é uma rara forma genética de neurofibromatose, recentemente descrita, na qual os doentes apresentam dois ou mais schwannomas não-vestibulares diagnosticados histologicamente. O diagnóstico implica a exclusão de neurofibromatose tipo 2 (NF2), com a qual partilha alguns aspectos em comum, através da realização de uma RMN crâneo-encefálica de alta resolução em doentes com mais de 18 anos de idade por forma a negar a existência de schwannomas vestibulares bilaterais. Isto é tanto mais importante porque o controlo evolutivo e o aconselhamento genético são muito diferentes nas duas entidades. Os autores apresentam um caso de schwannomatose múltipla não relacionada com NF2, incluindo um tumor no antebraço esquerdo,outro na parede abdominal e um maior no pé direito. A raridade da situação merece a divulgação deste caso.
Palavras chave: Schwannomatose, schwannomatose familiar, schwannomas múltiplos, schwannomas do pé.
ABSTRACT
Schwannomatosis is an uncommon recently described genetic form of neurofibromatosis, in which patients harbor two or more pathologically sampled non-vestibular schwannomas. Differential diagnosis includes neurofibromatosis type 2 (NF2), with which it shares some clinical aspects, making it mandatory to rule out bilateral vestibular schwannomas by performing a high-resolution MRI of the head (in patients over 18 years old). This is all the more important because follow-up and genetic counseling are vastly different in schwannomatosis and NF2. The authors present a case of NF2-unrelated multiple schwannomatosis in an 83-year-old woman with tumors in her left forearm, in her abdominal wall and a larger one in her right foot. This report aims to disclose this rare event and shed light on an obscure disease.
Key words: Schwannomatosis, familial schwannomatosis, multiple schwannomas, pedal schwannoma.
INTRODUÇÃO
Schwannomas são tumores raros que derivam das células de Schwann, células gliais que participam na bainha de mielina envolvendo o axónio e revestindo-se de tecido conjuntivo denominado epinervo[1,2]. Embora benignos, podem atingir grandes dimensões acabando por causar compressão nervosa[3]. São lesões de crescimento lento e indolor, frequentemente evoluindo ao longo de vários anos até ao diagnóstico mas podendo provocar fadiga muscular, parestesias ou dor crónica[4].
Estes tumores tipicamente ocorrem de forma isolada em indivíduos saudáveis. Schwannomas múltiplos do sistema nervoso periférico – nervos cranianos, raizes medulares, plexo braquial ou lombossagrado, ou nervos periféricos principais – estão descritos em associação com neurofibromatose tipo 2 (NF2) e outras síndromes raras como o complexo de Carney. Doentes jovens com NF2 tipicamente desenvolvem schwannomas vestibulares (SV) bilaterais e têm uma incidência aumentada de meningioma, ependimoma e astrocitoma, juntamente com diversas alterações oftalmológicas como opacidades lenticulares subcapsulares posteriores juvenis / cataratas corticais juvenis[4].
Nas duas últimas décadas surgiram relatos de doentes com schwannomas múltiplos e sem NF2 (sem SV bilaterais) – uma entidade nosológica definida como schwannomatose, considerada como uma terceira forma de neurofibromatose[5]. Existem duas formas de schwannomatose não relacionadas com NF2 – schwannomatose esporádica (SE) e schwannomatose familiar (SF). SE apresenta uma incidência de 1 novo caso para 1,7 milhões, semelhante à NF2, havendo apenas descrições raras de clusters familiares[6]. Ao invés a SF parece ter transmissão autossómica dominante. A schwannomatose múltipla exibe sintomas habitualmente a partir da terceira década, sendo a dor – agravando ao longo do tempo – o principal sintoma da maioria dos doentes5.
Os schwannomas do pé são raros [7,8] , mas quando presentes a sua localização mais comum é nos tecidos profundos[9].
CASO CLÍNICO
Doente de 83 anos, sexo feminino, admitida no serviço de Medicina a 20 de Maio de 2011 por pielonefrite aguda e traqueobronquite aguda. História prévia de angina estável, síndrome demencial e glomus timpânico tratado com radioterapia noutro hospital há 15 anos sem recidiva. Sem filhos; um irmão falecido com tumor cerebral agressivo, aos 65 anos, apenas 3 meses após os primeiros sintomas.
À admissão eram visíveis três massas: a primeira no pé direito, existente há mais de 15 anos; a segunda no antebraço esquerdo, presente há 5 anos; e a terceira na zona púbica, desde há 2 anos. Todas as lesões haviam crescido ao longo dos tempos e o tumor do pé era doloroso e limitativo da marcha há mais de 2 anos.
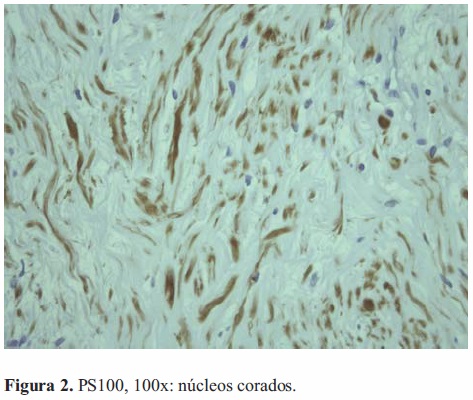
O exame mostrava lesões moles e bem-definidas. A do antebraço era ovóide e livre. A massa púbica era redonda e móvel. No pé o tumor era irregular e aparentemente fixo à fascia subjacente (Figura 1). Sem alterações na pele circundante, em qualquer um dos casos. Ecografia abdominal revelou imagem púbica nodular, com hipoecogenicidade heterogénea, margens bemdefinidas e estrutura vascular predominantemente periférica. Biopsias ecoguiadas às lesões do antebraço e da púbis evidenciaram schwannoma, com proliferação de células em paliçada e diferenciação neuronal (Vim+, PS100+, Actina-, Desmina-, CAM5.2-) (Figura 2).
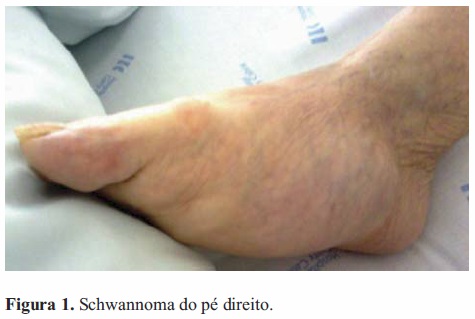
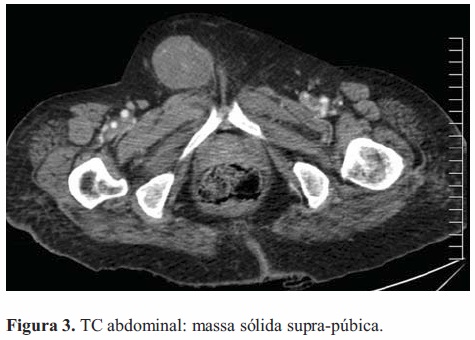
Foram efectuados diversos exames com vista ao esclarecimento das lesões. TC do antebraço revelou massa oval (56x36x27mm) com margens bem-definidas e em provável relação com o nervo mediano, limitada pela aponevrose muscular e sem sinais de ossificação. TC abdominal mostrou nódulo sólido púbico subcutâneo (51x44mm), em ligação com o nervo ilio-inguinal (Figura 3). TC do pé direito exibiu tumor capsulado bemdefinido, situado internamente na face plantar entre o calcâneo e as falanges. A cápsula era hiperdensa com conteúdo hipodenso, e a lesão comprimia as estruturas adjacentes.
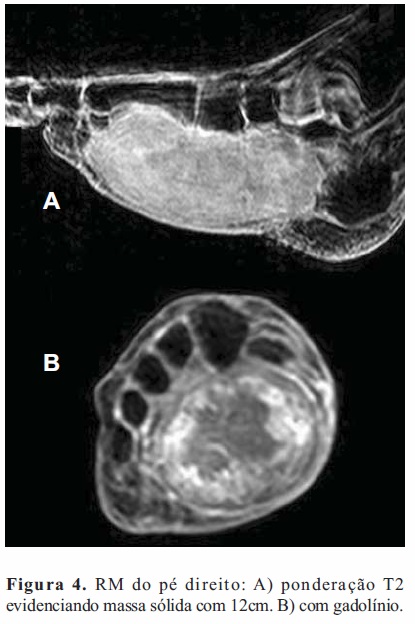
RM do pé direito confirmou massa sólida capsulada (120x63x54mm) na face plantar, desde a região perimaleolar interna, junto do tendão tibial posterior e dos tendões flexores dos dedos, isointensa em T1 e fortemente hiperintensa em T2 (Figura 4). Com gadolínio verificou-se acentuada captação periférica e uma zona central hipocaptante sugestiva de necrose (Figura 4). Sem alterações ósseas ou musculares.
TC crâneo-encefálica (CE) não evidenciou tumor vestibular, permitindo excluir NF2. Não foi considerada relevante a realização de RM CE.
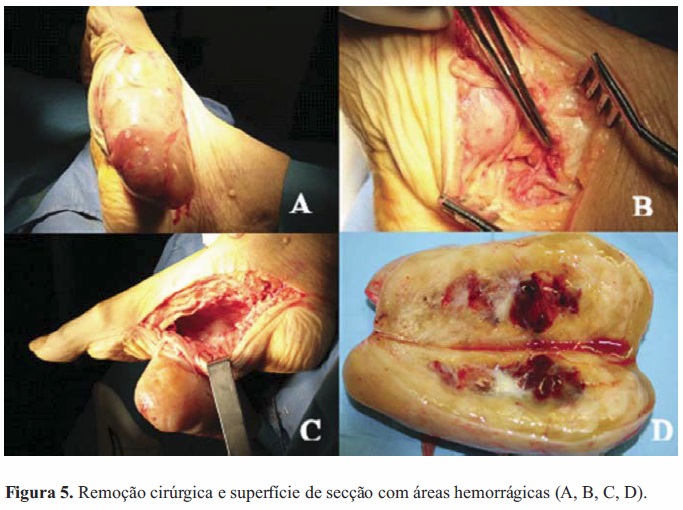
Atendendo à limitação na marcha, provocada pela dor, decidiu-se com a família pela ressecção cirúrgica da lesão no pé, intervenção decorrida de forma linear durante sessenta minutos e sem hemorragia significativa. A peça era creme com focos amarelados (Figura 5-A,B,C,D). Histologicamente apresentava cápsula e células fusiformes com áreas densamente celulares alternando com zonas de esparsa matriz disposta em paralelo, em paliçadas ou remoinhos. Núcleos grandes e pleomórficos, com escassas mitoses. A imunohistoquímica era positiva para vimentina e PS100, e negativa para actina, desmina e CD117.
Cinco dias após a cirurgia, a doente referiu febre e tosse com expectoração mucopurulenta, tendo iniciado empiricamente amoxicilina + ácido clavulânico endovenoso (ev). A cultura das secreções revelou Acinetobacter baumannii sensível a amicacina, colistina e tigeciclina, pelo que se mudou terapêutica para colistina ev. Apesar de alguma melhoria inicial, registou-se subsequente agravamento paulatino, com febre alta e refractária.
Instituiu-se vancomicina ev. Novas culturas de secreções brônquicas evidenciaram dois agentes: Escherichia coli produtora de beta-lactamases de espectro estendido (sensível a gentamicina e meropenem) e Staphylococcus aureus meticilino-resistente (sensível a gentamicina, cotrimoxazol e vancomicina). Adicionou-se meropenem ev mas o quadro evoluiu desfavoravelmente, falecendo a doente dois dias depois. Não foi requisitada autópsia.
DISCUSSÃO
Os schwannomas podem surgir em qualquer idade, mas mais frequentemente entre os 20 e os 60 anos.
Representam 5% das neoplasias benignas de tecidos moles[2]. Crescem lentamente e não apresentam variação por sexo ou raça[2]. Habitualmente decorrem meses a anos entre o início dos sintomas e a cirurgia curativa[2].
As topografias mais típicas são: superfícies flexoras das extremidades, pescoço, mediastino, retroperitoneu, raizes medulares posteriores e ângulo pontocerebeloso[2]. O aparecimento de queixas relaciona-se mais com o local do que com as dimensões do tumor, resultando da compressão nervosa que este provoca. O nervo implicado revela-se à periferia, achatado ao longo da cápsula mas sem penetrar a substância tumoral[2]. À observação as lesões são móveis transversalmente ao percurso do nervo mas fixas no plano longitudinal.
Macroscopicamente são massas globosas, tipicamente abaixo dos 5cm e rodeadas por cápsula de epinervo.
Seccionadas, revelam mistura de tecido fibroso acinzentado e focos amarelos; os tumores de maiores dimensões apresentam com frequência regiões quísticas[2]. Reconhecem-se dois padrões microscópicos: 1. áreas A de Antoni, que preenchem quase a totalidade dos schwannomas pequenos, com muitas células fusiformes organizadas em paliçada; 2. áreas B de Antoni, onde as células tumorais estão imersas em fluido abundante que pode formar espaços quísticos. As lesões antigas exibem também células isoladas com núcleos hipercromáticos bizarros, sem significado patológico. Há escassas mitoses. Por vezes a existência de muitos vasos pode simular neoplasia vascularizada. Podem aparecer também fibras “amiantóides” ou esferas de colagénio com bordos espiculados, e os maiores tumores podem ter macrófagos esponjosos2. Raramente surgem áreas epitelióides ou plexiformes[2].
A imunohistoquímica é positiva para proteína S100, mielina, calretinina, calcineurina, componentes da lâmina basal, vimentina, receptor para o factor de crescimento do nervo e lipocortina-1[2].
A TC mostra uma massa bem-definida, iso- a hipointensa em relação ao músculo e captando contraste. A RM é o método de imagem mais útil no diagnóstico, mostrando: 1) em T1, uma intensidade igual ou ligeiramente maior do que a do músculo; 2) em T2 ou T1 contrastado, padrão em alvo com halo periférico hiperintenso e baixa intensidade central. Estes aspectos surgem também em tumores malignos de tecidos moles, cuja imagem por RM pode ser semelhante à dos schwannomas[10].
O diagnóstico definitivo de schwannomatose aguarda identificação do locus exacto. Para melhor organização clínica e de pesquisa, a National Neurofibromatosis Foundation propôs um Consenso para uniformizar critérios diagnósticos[4], dividindo os casos em “confirmados”, “prováveis” e “segmentares”.
Schwannomatose ”confirmada” pode englobar: A) idade >30 anos, >2 schwannomas não--intradérmicos confirmados histologicamente, sem evidência de tumor vestibular por RM de alta resolução, e sem mutações em NF2, ou B) um schwannoma não-vestibular com confirmação histológica e um parente directo que preencha o primeiro critério. Schwannomatose “provável” compreende: A) idade <30 anos, >2 schwannomas não intradérmicos, pelo menos um com confirmação histológica, sem evidência de tumor vestibular em RM de alta definição e sem mutações em NF2; ou B) idade >45 anos, >2 schwannomas não intradérmicos, pelo menos um com confirmação histológica, sem sintomas de disfunção vestibulococlear, sem mutações em NF2 ou evidência radiográfica de schwannoma não-vestibular e com um parente directo que preencha o primeiro critério para schwannomatose confirmada. Schwannomatose ”segmentar” ou “em mosaico” limita-se a um membro ou a <5 segmentos vertebrais contíguos.
Detectam-se mutações constitucionais do gene NF2 apenas em 65% dos novos casos de NF2, e esta patologia é francamente improvável em alguém acima dos 45 anos de idade e sem sintomas vestibulococleares[11].
Na investigação de um doente, a exclusão de NF2 em casos duvidosos obriga à realização de RM de alta resolução do sistema nervoso central (crâneo-encefálica e da coluna vertebral), exame oftalmológico detalhado e teste genético para mutações constitucionais de NF2[11]. SV não estão presentes na schwannomatose. Contudo, a incidência de SV unilaterais esporádicos aumenta acima dos 50 anos, pelo que a presença de SV unilaterais num doente não implica que estejam relacionados com outros schwannomas periféricos. O mesmo vale para doentes com schwannomas múltiplos não-vestibulares que desenvolvam meningiomas ou cataratas em idade mais avançada. Inversamente, a suspeita deve pender para NF2 (e não schwannomatose) quando surgem cataratas, schwannomas cutâneos, meningiomas, gliomas ou ependimomas em doentes jovens com schwannomas múltiplos não-vestibulares[11].
Embora possa haver sobreposição clínica e fenotípica entre schwannomatose e NF2[4], estas são entidades distintas. Estudos moleculares revelam presença, nos doentes com NF2, de mutação germinativa autossómica no gene NF2 em 22q12[12], enquanto a schwannomatose demonstra linkage não-germinativo ao cromossoma 22[12]. Há relatos de alterações no locus do gene NF2 em tumores associados a schwannomatose, mas nunca em tecidos não-tumorais. O locus da schwannomatose não foi ainda identificado mas estudos de linkage delimitamno a um intervalo com 5 milhões de pares de bases, proximal ao gene NF2 no cromossoma 22[4].
Foram encontradas mutações germinativas constitucionais no gene oncossupressor INI1 como causa em 30-60% dos casos de SF e em menor percentagem na SE[13,14].
A principal indicação cirúrgica dos schwannomas continua a ser a dor refractá ria. O tratamento indicado é a remoção extra- ou intracapsular. A dissecção meticulosa dos schwannomas consegue remover completamente o tumor preservando o nervo. Frequentemente é necessário recorrer à ampliação microscópica ou por lupa, para não danificar fibras nervosas durante dissecção epi- e endoneural, evitando assim perda neurológica. A recorrência do schwannoma após excisão cirúrgica é rara[15].
REFERÊNCIAS BIBLIOGRÁFICAS
1. Huang JH, Simon SL, Nagpal S, Nelson PT, Zager EL. Management of patients with schwannomatosis: report of six cases and review of the literature. Surg Neurol. 2004; 62: 353-361 [ Links ]
2. J Rosai. Schwannoma (neurilemmoma). In Rosai J, editors. Rosai and Ackermans Surgical Pathology. New York: Mosby; 2011. p. : 2130-2132.
3. Ogose A, Hotta T, Morita T, Otsuka H, Hirata Y. Multiple schwannomas in the peripheral nerves. J Bone Joint Surg (Br). 1998; 80: 657-661 [ Links ]
4. MacCollin M, Chiocca EA, Evans DG. Diagnostic criteria for schwannomatosis. Neurology. 2005; 2005 (64): 1838-1845 [ Links ]
5. MacCollin M, Woodfin W, Kronn D, Short MP. Schwannomatosis: a clinical and pathologic study. Neurology. 1996; 46: 1072-1079 [ Links ]
6. MacCollin M, Willett C, Heinrich B. Familial schwannomatosis: exclusion of the NF2 locus as the germline event. Neurology. 2003; 60: 1968-1970 [ Links ]
7. Mangrulkar VH, Brunetti VA, Gould ES, Howell N. Unusually large pedal schwannoma. J Foot Ankle Surg. 2007; 46: 398-402 [ Links ]
8. Joyce M, Laing AJ, Mullet H. Multiple schwannomas of the posterior tibial nerve. Foot Ankle Surgery. 2002; 8: 101-103 [ Links ]
9. Mendeszoon MJ, Cunningham N, Crockett RS, Kushner D. Schwannoma: a case report. Foot and Ankle Online J. 2009; 2 (10): 4 [ Links ]
10. Varma DG, Moulopoulos A, Sara AS. MR imaging of extracranial nerve sheath tumours. J Comput Assist Tomogr . 1992; 16: 448-453 [ Links ]
11. Baser ME, Friedman JM, Evans DG. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006; 66: 730-732 [ Links ]
12. Westhout FD, Mathews M, Paré LS, Armstrong WB, Tully P, Linskey ME. Recognizing schwannomatosis and distinguishing it from neurofibromatosis type 1 or 2. J Spinal Disord Tech. 2007; 20: 329-332 [ Links ]
13. Hadfield KD, Newman WG, Bowers NL. Molecular characterization of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet. 2008; 45: 332-339 [ Links ]
14. Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007; 80: 805-810 [ Links ]
15. Ozdemir O, Ozsoy MH, Kurt C, Coskunol E, Calli I. Schwannomas of the hand and wrist: long-term results and review of the literature. J Orthop Surg (Hong Kong). 2005; 13: 267-272 [ Links ]
Conflito de interesse:
Nada a declarar.
Antonio Murinello
Avª Engº Antº Azevedo Coutinho, Lte 8 r/c – dto
2750 Cascais
Portugal
amurinello@gmail.com
Data de Submissão: 2012-08-09
Data de Revisão: 2012-10-15
Data de Aceitação: 2012-10-30


{kind=link}