











 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntrodução
Os linfomas são um grupo heterogéneo de neoplasias malignas caracterizado pela proliferação de linfócitos clonais nos gânglios linfáticos e outros órgãos linfóides (baço, fígado), podendo também infiltrar a medula óssea e outros órgãos não linfoides.
Os linfomas não‑hodgkin (LNH), são os linfomas mais frequentes, correspondendo a cerca de 4‑5% de todos os tumores malignos. Os LNH compreendem um grupo de neoplasias malignas de células B, T ou NK em vários estádios de diferenciação, 90% destes de célula madura. Uma significante proporção de doentes com LNH (20‑30%),
têm envolvimento extra‑ganglionar.1 A região de cabeça e pescoço é o segundo local mais comum de manifestação extra‑ganglionar, sendo mais comum o seu aparecimento no anel de Waldeyer.2 A cavidade oral é uma localização incomum LNH com uma incidência de 0,1 - 5 %.1,3
O linfoma folicular (LF) é o segundo subtipo mais comum de LNH, representando cerca de 20% de todos os linfomas.4 O LF afeta predominantemente a população adulta, apresentando maior incidência entre a sexta década de vida, com ligeiro predomínio no género feminino.5
O LF é uma neoplasia derivada das células B do folículo central (tipicamente centrócitos e centroblastos) e que apresenta habitualmente um padrão folicular, dando uma aparência nodular macroscópica e microscopicamente.5
Relativamente à imunohistoquímica as células tumorais expressam antigénios associados às células B (CD19, CD20, CD22, CD79a) e são usualmente positivos para BCL2, BCL6 e CD10 e negativos para CD5 e CD43.6
Pode ser classificado quanto ao grau consoante o número de centroblastos por campo de alta potência: grau 1 (≤5), grau 2 (6‑15) e grau 3 (>15). LF de grau 1‑ 2 são tipicamente linfomas de baixo grau de proliferação, enquanto grau 3 podem manifestar‑se com maior agressividade.7
O LF está tipicamente associado à translocação cromossómica t(14;18)(q32;q21), entre o gene IGH e BCL2. Esta translocação resulta na sobre‑expressão da proteína anti‑apóptica intracelular BCL‑2 que permite que as células B neoplásicas inibam o processo apóptico programado, apresentando desta forma, sobrevida prolongada.8 A translocação t(14;18) (q32;q21) é identificada em até 85‑90% dos LF grau 1‑2.9
Relativamente ao curso clínico, o LF corresponde ao mais comum linfoma indolente. Os linfomas indolentes geralmente são incuráveis com as abordagens terapêuticas standard e são caracterizados por um curso crónico com repetidas recidivas; no entanto, muitos doentes sobrevivem largos anos com doença estável, na ausência de tratamento específico.
O linfoma folicular envolve predominantemente os gânglios linfáticos, assim como baço, medula óssea, sangue periférico e menos comumente o anel Waldeyer. Qualquer grupo de gânglios linfáticos pode estar envolvido e a maioria dos doentes apresentam adenopatias periféricas. Apresentações puramente extra‑ganglionares são extremamente raras.
O linfoma folicular oral (LFO) apresenta pouca informação clínica, epidemiológica e patológica documentada.10
Os linfomas com envolvimento extra‑ganglionar que se manifestam na cavidade oral compreendem menos de 5% de todas as neoplasias que afetam a cavidade oral, sendo que a grande maioria surge predominantemente no palato duro, seguido da língua.11) A maioria dos linfomas relatados na literatura (68%) que se manifestam na cavidade oral correspondem a linfoma B difuso de grandes células (LBDGC).12
Caso clínico
É descrito um caso clínico de uma doente de 59 anos, género feminino, sem antecedentes pessoais relevantes, referenciada para a consulta externa do Serviço de Estomatologia do Hospital Geral dos Covões/ Centro Hospitalar Universitário de Coimbra (CHUC), com queixas de edema na região do palato com cerca de dois meses de evolução. A doente referiu que a lesão se apresentava indolor, de crescimento gradual. Sem outra sintomatologia associada, nomeadamente sintomas constitucionais.
O exame físico revelou um abaulamento assimétrico do hemi‑palato duro esquerdo revelando uma lesão dura, edemaciada e restrita à região edemaciada (Figuras 1 e 2).
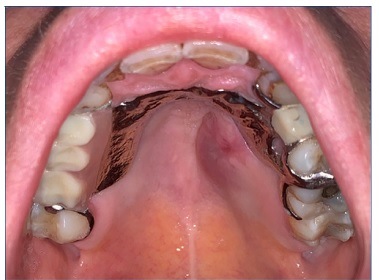
Figura 1 Lesão intra‑oral localizada no hemi‑palato duro (com prótese removível esquelética colocada)
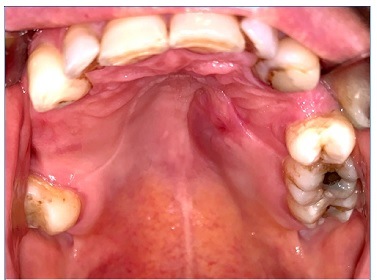
Figura 2 Lesão intra‑ oral localizada no hemi‑palato duro (sem prótese removível esquelética colocada)
A ortopantomografia não revelou qualquer envolvimento dos tecidos ósseos (Figura 3).
As hipóteses colocadas para diagnóstico diferencial desta lesão apresentadas foram tumores de células salivares (adenoma pleomórfico, carcinoma muco‑epidermoide e adenocarcinoma polimórfico de baixo grau), abcesso de origem dentária/endôntica ou de origem periodontal, e neoplasia linfoproliferativa. Deste modo, procedeu‑se à biópsia incisional que revelou, a nível submucoso, a existência de glândulas mucosas rodeadas por denso infiltrado de células linfoides com vago padrão nodular, assim como espessamento hialino acentuado periductal (Figura 4). Os linfócitos apresentavam tamanho pequeno a intermédio com núcleos hipercromáticos e irregulares, sem lesões linfoepiteliais. A análise imunohistoquímica, revelou expressão de CD20 generalizada com CD10 positivo em áreas nodulares, assim como BCL‑2 e BCL‑6 (Figuras 5, 6 e 7). Estas alterações eram compatíveis com linfoma B folicular, com envolvimento de glândulas salivares minor.
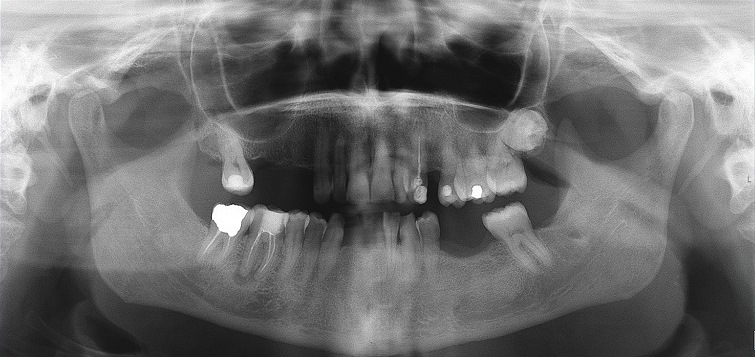
Figura 3 Ortopantomografia no momento da avaliação demonstrando ausência de envolvimento ósseo da lesão
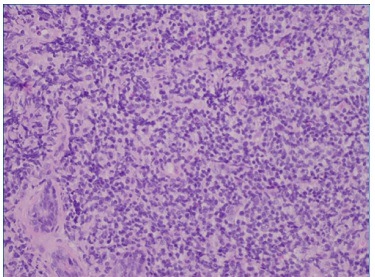
Figura 4 Anatomia patológica da biópsia incisional, revelando existência de glândulas mucosas rodeadas por denso infiltrado de células linfoides com vago padrão nodular

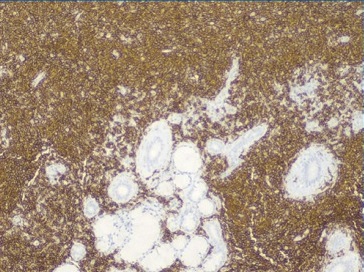
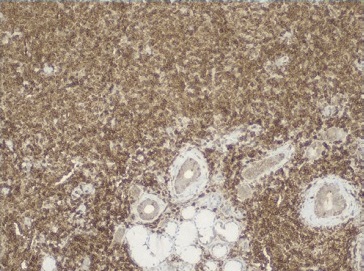
Dado o diagnóstico em causa, procedeu‑se ao pedido de colaboração do Serviço de Hematologia do CHUC que deu seguimento ao estudo, estadiamento e tratamento da patologia.
Do estudo analítico realizado, não apresentava alterações, nomeadamente linfocitose ou citopenias ou aumento da LDH. A imunofenotipagem do aspirado medular que revelou a presença de 1,1% de linfócitos com fenótipo sugestivo de LF. Não foi possível confirmar a t(14;18) por FISH, uma vez que as células eram escassas. A biópsia óssea demonstrou envolvimento nodular paratrabecular por linfoma não‑Hodgkin B, cujo padrão folicular sugeria LF.
Foi efetuado o estudo por tomografia computorizada (TC) cervical, torácica e abdómino‑pélvico (Figura 8), que identificou adenopatias nas cadeias linfáticas supra e infradiafragmáticas nível as maiores ao longo dos compartimentos mediastínicos, a maior com 36‑300mm; cadeias lombo‑aórticas (35x20 mm) e regiões ilíacas (49x29 mm).
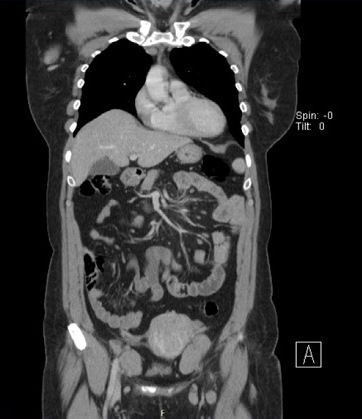
A doente iniciou tratamento com imuno‑quimioterapia segundo o protocolo R‑CHOP (rituximab, ciclofosfamida, doxorrubicina, vincristina e prednisolona), durante 6 ciclos. No fim do 3.º R‑CHOP foi possível observar a total regressão da lesão do palato (Figuras 9 e 10). A doente completa os 6 ciclos de quimioterapia com resposta completa e prossegue tratamento com rituximab de manutenção a cada 2 meses, durante 2 anos.
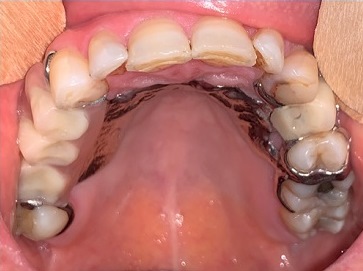
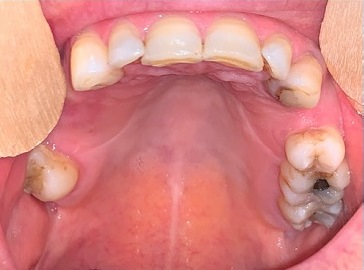
Figura 9 Regressão total da lesão anteriormente observada hemi‑palato (com prótese removível esquelética)
Discussão e conclusões
A afeção do palato no linfoma folicular é rara.13 O LF é observado em 6% dos casos,14 e a transformação deste para um subtipo histologicamente agressivo (alto grau), como linfoma difuso de grandes células B ocorre em 20‑30% dos casos.10
Muitos doentes são assintomáticos ao diagnóstico. As manifestações clínicas podem mesmo não existir, podendo na maioria das vezes manifestar‑se apenas por fadiga ou astenia prolongada. Noutros casos, poderá incluir febre, sudorese nocturna ou perda ponderal. Devido ao seu comportamento clínico indolente, a maioria dos doentes com LF apresenta doença disseminada no momento do diagnóstico inicial. Este tipo de linfoma geralmente é diagnosticado por biópsia excisional de um gânglio linfático. Biópsias de core também poderão ser usadas quando não há gânglios acessíveis. Biópsias por punção aspirativa de agulha fina não são adequadas. Mais raramente o diagnóstico é feito por biópsia de massa extra‑ganglionar, como foi o caso desta doente.
Neste caso, o diagnóstico diferencial incluiu tumores de células salivares (adenoma pleomórfico, carcinoma muco‑epidermoide e adenocarcinoma polimórfico de baixo grau) assim como abcesso periodontal e o linfoma não‑Hodgkin.15 O adenoma pleomórfico, é a neoplasia mais comum das glândulas salivares major e minor. O palato é local mais comum quando ocorre nas glândulas salivares minor. O carcinoma muco‑epidermoide é o tumor salivar maligno mais comum. Depois da glândula parótida, o palato é o local mais comum de manifestação deste tumor. Por ser o tumor maligno que atinge exclusivamente as glândulas salivares minor, especialmente no palato duro, o adenocarcinoma polimórfico de baixo grau também era um dos diagnósticos possíveis.16 Condições benignas como abcessos de origem dentária ou origem periodontal são hipóteses que também devem ser tidas em conta.17 Todas estas patologias têm capacidade de se manifestar por edema do palato apresentando um crescimento lento e assintomático, pelo que fizeram parte das hipóteses para o diagnóstico da lesão.
No que respeita ao estadiamento, a tomografia computadorizada (TC) é o exame de eleição no LF. O exame por TC deve incluir a região cervical, tórax, abdómen e pélvis. PET‑CT poderá ser recomendada em caso de suspeita de transformação para linfoma agressivo ou para confirmar doença localizada.
O estadiamento deverá ser complementado com biopsia óssea para verificar se há infiltração medular.18
Os doentes com LF são estadiados segundo a Classificação de Ann‑Arbor, de I a IV, consoante o grau de extensão da doença: localizada - estádios I‑II; disseminada - estádios III‑IV. Esta doente encontrava‑se num estádio IV de Ann Arbor.18
O tratamento irá depender do estadiamento da doença, do estado geral do doente e sinais de alta carga tumoral (critérios GELF (Groupe d’Etude des Lymphomes Folliculaires)). Na doença localizada a radioterapia (24 Gy) poderá ser potencialmente curativa. Rituximab em monoterapia poderá ser usado para evitar toxicidade da radioterapia. Em casos seleccionados vigilância ou quimioterapia poderão estar indicados. Já na doença disseminada não existe tratamento curativo. Só deverá ser iniciado tratamento se estiverem presentes sinais de alta carga tumoral: massas bulky (>7 cm) ou 3 regiões ganglionares com >3 cm; esplenomegalia sintomática; compressão de órgão pelo tumor, derrame pleural ou peritoneal; elevação de LDH ou beta2‑microglobulina; sintomas B (perda ponderal >10%, febre ou hipersudorese noturna). Na doença disseminada, o tratamento recomendado é a imuno‑quimioterapia, sendo as combinações com rituximab com CHOP ou Bendamustina as que alcançam maior sobrevivência livre de progressão. Esquemas menos intensivos poderão ser usados em doentes mais frágeis. A manutenção com Rituximab a cada 2 meses durante 2 anos aumenta a sobrevivência livre de progressão e está recomendada após tratamento com imuno‑quimioterapia.18
O prognóstico do LF encontra‑se muito relacionado com a extensão da doença no momento do diagnóstico, tendo sido desenvolvida uma escala de prognóstico específica - FLIPI (Follicular Lymphoma International Prognostic Index). Os 5 preditores de independentes de pior prognóstico são: idade >60 anos, hemoglobina <12 g/dL, elevada LDH, doença disseminada e >4 cadeias ganglionares envolvidas. A presença de 0‑1, 2, 3‑5 destes fatores, respetivamente define baixo‑risco, risco‑intermédio e alto risco. A sobrevivência global estimada ao 10 anos varia de 70% nos doentes de baixo risco para 35% nos doentes de alto risco (FLIPI). A doente do caso descrito apresentava um FLIPI2, que apresenta uma sobrevida estimada aos 10 anos de 50%.
Com os tratamentos atuais, a sobrevida mediana dos doentes com LF é superior a 12 anos. No entanto 25 a 35% dos doentes com LF ocorre transformação para LBDGC, linfoma agressivo e de prognóstico sombrio. Mais raramente poderá haver transformação noutro tipo de linfoma agressivo como: Linfoma de Burkitt, Linfoma de Hodgkin ou Linfoma linfoblástico B.19
Em suma, os tumores Não‑Hodgkin, apesar de raros na cavidade oral, devem ser sempre considerados no diagnóstico diferencial quando se suspeita de doenças malignas intra‑orais.
Pela sua raridade, todos os relatos de caso são importantes para cimentar o conhecimento existente. A identificação precoce e o encaminhamento imediato para o especialista diferenciado resultariam no manejo oportuno, e determinantes de um melhor prognóstico do doente.

