











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Neurofibromatosis 1 (NF1) is the most common type of neurofibromatosis (NF).1 The earliest clinical manifestations of NF1 are café-au-lait pigmentations, axillary freckling, and Lisch nodules. Other manifestations include skeletal deformities and optic glioma. Neurofibroma and plexiform neurofibroma tend to manifest in early adolescence.2
The malignant peripheral nerve sheath tumor (MPNST) is a rare type of sarcoma that accounts for 2-6% of all head and neck sarcomas.1 MPNST can be associated with NF1, which occurs in 50% of MPNST cases. In turn, MPNST is reported to develop in 8-12% of NF1 cases, mainly arising from a pre-existing plexiform neurofibroma.3 Le Guellec et al.4 reported that MPNST cases related to NF1 tend to occur in younger age groups and present as more rapidly growing masses than sporadic cases. Cai et al.5 evaluated the prognosis of MPNST cases in the English literature and found that cases associated with NF1 and cases in the head and neck region had a poorer prognosis and lower overall survival rate. This article reports a case of low-grade MPNST with rhabdoid features arising from a plexiform neurofibroma in the temporal region of a patient with NF1. It analyzes the management and prognosis of the case.
Case report
A 20-year-old male patient presented with a large well-circumscribed swelling that extended across the right temporal, zygomatic, and parotid regions, causing downward displacement of the auricle (Figures 1 and 2). The previous year, the patient had suffered from a slow-growing mass on the same site of the current lesion, which was diagnosed as a plexiform neurofibroma. The patient was diagnosed with NF1 in early childhood. He had numerous café-au-lait pigmentations with smooth borders on his arms, back, chest, and neck (Figures 1 and 2). Moreover, he had multiple nodules on his neck, back, and arms, diagnosed as neurofibromas whenever excised.
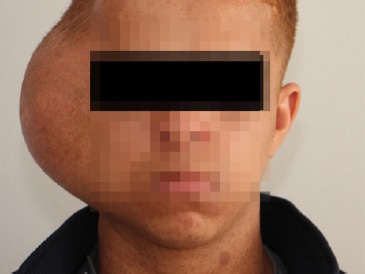
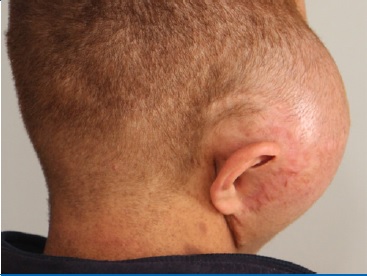
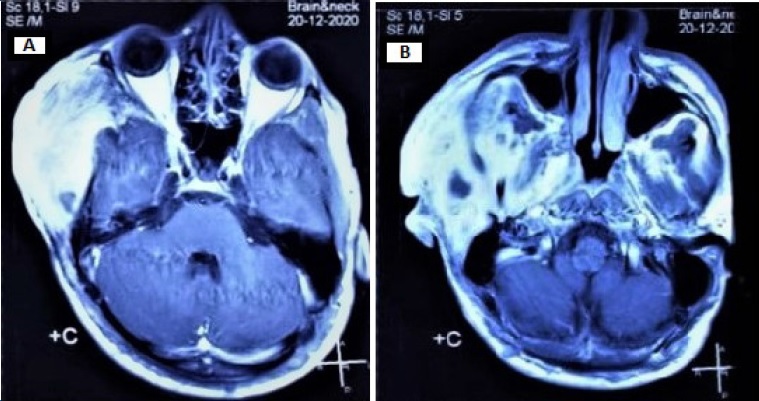
There was no family history of NF1. MRI examination revealed a well-defined subcutaneous mass involving the right side of the face and temple, with 12.5 x 8.8 x 5.8 cm (craniocaudal/ anteroposterior/ transverse), that showed a heterogeneous signal, being hypointense in T1W sequence and hyperintense in T2W. The mass caused a lower displacement of the right earlobe overlaying the ramus of the mandible and located superficial to the parotid gland. Furthermore, the lesion showed a deep extension into the right parapharyngeal space (Figure 3). The patient performed a PET scan that revealed he was free of any metastasis.

The lesion was approached through a modified Blair incision with temporal and cervical extensions (Figure 4). The temporal part of the lesion, which was removed through extracapsular dissection, was displacing the facial tissues rather than infiltrating and its deep surface was against the temporal boné (Figure 5). The parapharyngeal part was approached through the cervical extension of the incision and was removed by blunt finger dissection.
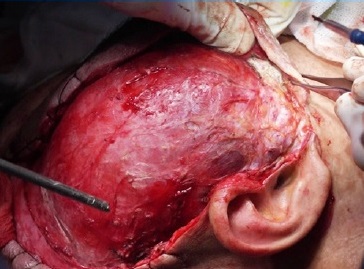
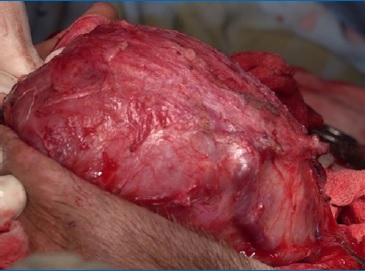
Figure 5 Intraoperative image: the temporal part of the lesion was displacing the facial tissues rather than infiltrating them.
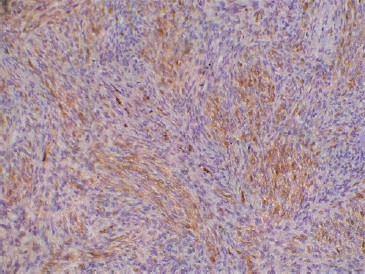
The excised lesion was sent to the pathology lab, which revealed hypercellular interlacing fascicles of spindle cells with hyperchromatic wavy thin nuclei. Cellular atypia and increased mitosis (≥5 mitosis/ 10 HPF) were evident in all fields (Figure 6). Some fields revealed ovoid cells with eosinophilic cytoplasm and others with vacuolated cytoplasm, giving the appearance of “spider cells” (Figures 6 C and D). Little necrosis and hemorrhage were detected. Immunohistochemical staining with S100 protein revealed patchy nuclear and cytoplasmic positivity (Figure 7).
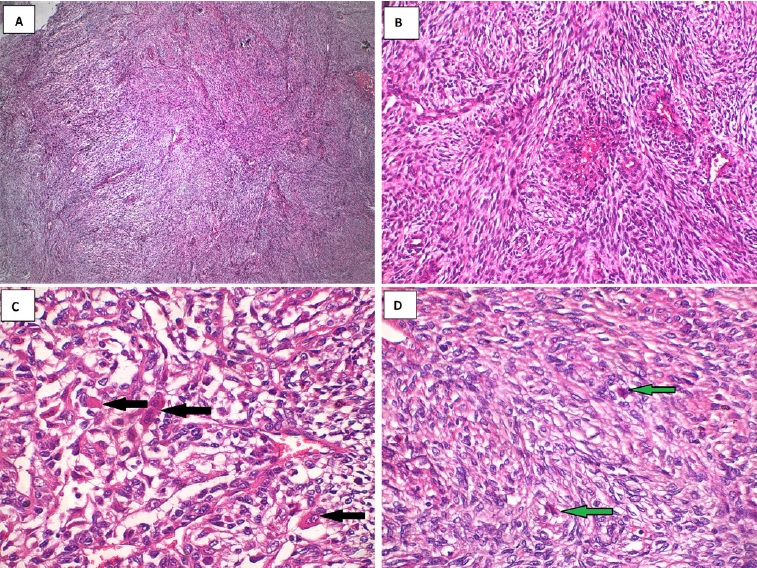
Figure 6 Histopathological findings: A. Marbled appearance due to alternating hypocellular and hypercellular areas; B. Perivascular accentuation and evident herringbone growth pattern; C. Increased mitosis and rhabdoid cells with eosinophilic cytoplasm (black arrows); D. Rhabdoid cells with vacuolated cytoplasm (“spider cells” green arrows).
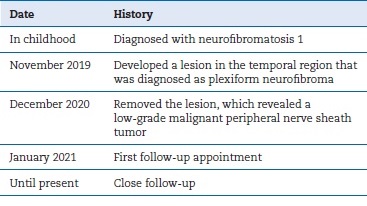
Based on the histopathological and immunohistochemical staining, along with the history of the lesion, the lesion was diagnosed as a low-grade MPNST. Two weeks after the operation, the patient came for follow-up (Figure 8), and the MRI scan revealed only a considerable amount of post-surgical edema (Figure 9). The patient started high precision intensity- modulated radiotherapy (IMRT) up to 60 Gy. The patient has been under ongoing close follow-up for the last 6 months. He did not report complaints during the check-ups. The exact timeline of the case can be seen in Table 1.
Discussion and conclusions
Generally, NF1 is considered one of the most prevalent autosomal dominant genetic conditions affecting human beings.6 The pathogenesis of NF1 is based on the loss of function mutation that occurs in the neurofibromin 1 gene, located on chromosome 17q11.2 Normally, neurofibromin serves as a tumor suppressor protein, responsible for activating the GTPase-activating protein, which leads to the inactivation of Ras and inhibition of cell proliferation.2 However, half of the NF1 cases result from spontaneous mutations, as in our case.2 Extra attention should be given to the plexiform variant of neurofibroma, which is regarded as a precursor for MPNST, as occurred in the present case.2,3
Miettinen et al.6 7 proposed nomenclature for NF1-associated lesions. The first entity, the atypical neurofibromatous neoplasm of uncertain biologic potential (ANNUBP), has at least two of the following features: nuclear atypia, hypercellularity, loss of neurofibroma architecture, and/or mitotic activity >1/ 50 HPFs and <3/ 10 HPFs. On the other hand, low-grade MPNST shows no necrosis and has a mitotic index of 3- 9/10 HPFs. In turn, high-grade MPNST is characterized by necrotic areas and a high mitotic index of >10/ 10 HPFs. According to the above-mention scheme, our case fulfilled the criteria of low-grade MPNST.
In our case, there was focal positivity for S-100 staining, although S-100 may show decreased sensitivity for MPNST.8 The current most specific marker of MPNST is the loss of H3K27me3 (trimethylated histone 3 at lysine residue 27), which has been reported to occur in 61% of MPNST cases, including those associated with NF1. It is also useful in differentiating between ANNUBP and MPNST as it is not lost in ANNUBP.8 Since our case showed positivity to the S-100 protein and was associated with NF1, we considered there was no need to do the H3K27me3 staining.
In the literature, the MPNST containing cells with rhabdoid features was called malignant triton tumor (MTT) and is a rare subtype of MPNST. Some authors suggested that the origin of the neural cells and rhabdoid cells in MPNST is undifferentiated neural crest cells.9 Based on the previously reported cases, MTT was postulated as having a highly aggressive clinical behavior and a low 5-year survival rate.9
In general, when MPNST is compared to other soft-tissue sarcomas, it is considered to have a worse prognosis, with up to 65% local recurrence rate and up to 68% distant metastasis.
Several prognostic factors such as tumor size, tumor site, histopathological grade, and involvement of surgical margins have been suggested, but no consensus has been reached.10
There is a conflict in the literature on whether MPNST’s association with NF1 and occurrence in the head and neck region lead to a worse prognosis.5 In our case, the close follow-up of the NF1 diagnosed patient led to the early diagnosis of the malignancy. Moreover, although our case was in the temporal region, it revealed a low-grade lesion, contradicting the opinion of the worse prognosis of head and neck lesions. Due to the irresponsive feature of the tumor to definitive chemoradiotherapy, the treatment of choice is surgical resection with wide safety margins, along with adjuvant radiation therapy for large tumors.5
We conclude that the prognosis of MPNST or MTT is multifactorial and cannot be determined by just one prognostic factor. Furthermore, the association of MPNST with NF1 may lead to an early diagnosis of the lesion due to the close follow-up.

