











 Portugués (pdf)
Portugués (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntrodução
As malformações do sistema urinário são as anomalias mais frequentemente diagnosticadas no período pré-natal (20-30%)1,2,3. O seu espetro é amplo, desde malformações assintomáticas até situações graves, incompatíveis com a vida4. O diagnóstico pré-natal é essencial para o tratamento precoce e prevenção da deterioração da função renal.
O presente artigo tem como objetivo rever as malformações mais frequentes do sistema urinário, com ênfase sobre os achados ecográficos e diagnóstico diferencial, e propor um protocolo de atuação clínica.
Para efeitos desta revisão, as anomalias serão classificadas em: anomalias de número, anomalias de posição/fusão, anomalias do parênquima renal e dilatações do sistema coletor.
Avaliação ecográfica do sistema urinário fetal
Em Portugal, é realizada por rotina uma ecografia no 1º trimestre (11-13+6 semanas). Nesta fase, a bexiga apresenta estrutura anecóica com paredes hiperecogénicas, circundada pelas artérias umbilicais, que podem ser visualizadas com color Doppler (figura 1-A)5. Os rins poderão eventualmente já ser visíveis5.
Esta avaliação inicial é posteriormente com-plementada com a ecografia morfológica (20-22 semanas). Na avaliação dos rins, podemos ver uma zona externa e hiperecogénica, o córtex, e uma zona interna e hipoecogénica, a medula. No plano axial, os rins apresentam-se como estruturas arredondadas na região paravertebral, sendo este o plano da medição da largura e espessura dos rins (figura 1-B). A medição do comprimento renal é realizada no plano coronal, no qual os rins apresentam forma elíptica (figura 1-C), com as glândulas suprarrenais visíveis cranialmente5.
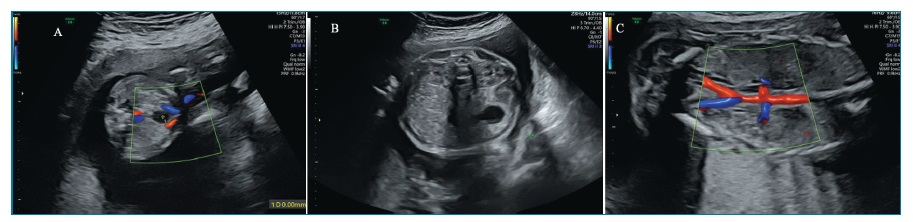
Figura 1 Aparência ecográfica normal dos rins, artérias renais e bexiga. A- Artérias umbilicais a circundar a bexiga (cursor) com Doppler a cores; B - Visualização dos rins na região paravertebral - Plano Axial; C - Visualização dos rins e artérias renais com Doppler a cores - Plano Coronal
Os ureteres e a uretra habitualmente não são visíveis na ecografia, exceto quando dilatados5.
A partir das 16-17 semanas, o líquido amniótico é produzido quase exclusivamente através da urina fetal, pelo que a presença de oligoâmnios/anidrâmnios a partir desta idade gestacional requer sempre investigação5,6.
Anomalias de número
Agenesia renal
Corresponde à ausência congénita de um ou ambos os rins1.
A sua prevalência é de 1:2000 (unilateral) e de 1:5000 nascimentos (bilateral). É mais frequente no sexo masculino e à esquerda7,8.
Ecograficamente, na AR unilateral não se visualiza um dos rins (figura 2-A), sendo a bexiga e o volume de líquido amniótico (VLA) normais. O rim único pode encontrar-se aumentado de tamanho em 90% dos fetos9,10. Uma razão entre os diâmetros anteroposterior e transverso do rim superior a 0,9 sugere hipertrofia renal compensatória9. O Doppler a cores demonstra uma única artéria renal (figura 2-B). A bilateral caracteriza-se por ausência de visualização de rins, bexiga e das artérias renais e anidrâmnios (a partir das 17 semanas). As glândulas suprarrenais assumem uma forma discoide e movem-se lateral e inferiormente11,12. Como exame complementar para despiste de outras malformações com oligo/anidrâmnios, poderá realizar-se ressonância magnética (RM) fetal13.
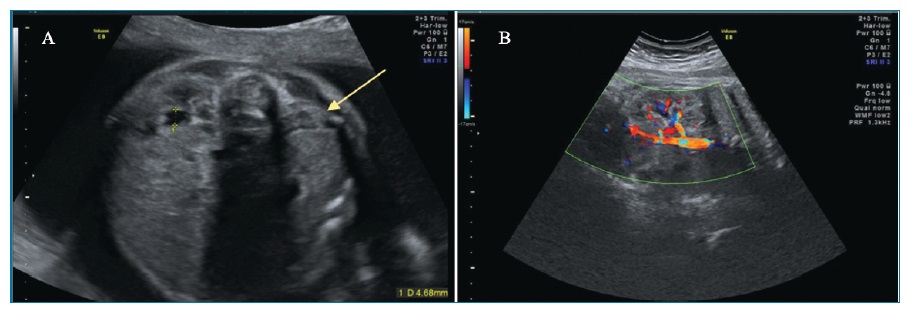
Figura 2 A- Agenesia renal esquerda com glândula suprarrenal a preencher a fossa renal (seta). B- Artéria renal esquerda visível e ausência de visualização da artéria renal direita no Doppler a cores
Na maioria dos casos, é uma anomalia isolada esporádica, estando em 20-30% dos casos associada a Síndrome de Fraser, associação VACTERL, associação MURCS, síndrome de regressão caudal, síndrome de Rokitansky-Kuster-Hauser, entre outros5.
Perante uma AR, é importante pesquisar malformações adicionais do rim contralateral, genitourinárias, musculoesqueléticas (40%) ou cardiovasculares (15%). Se presentes, poderá ser realizado cariótipo/array-CGH. Nos fetos do sexo feminino, deverá ser pesquisada a presença de malformações Müllerianas 5,14. Em 1-2% dos casos associa-se a alterações cromossómicas, sendo a mais frequente a trissomia 18.
A AR bilateral pode cursar com morte fetal in utero em até 33% dos casos15. Como resultado do oligoâmnios precoce grave, surgem eventos disruptivos designados de sequência de Potter (hipoplasia pulmonar, alterações na face e pele e deformações dos membros), sendo lícito o casal ponderar interrupção médica da gravidez (IMG)5. Não há evidência científica robusta que apoie a realização de amnioinfusões seriadas, implementadas apenas em contexto investigacional4,16.
No caso da AR unilateral isolada, não há indicação para ecografias seriadas, dependendo apenas de critérios obstétricos. O prognóstico é bom, quando o rim contralateral não se encontra afetado. Em cerca de 20% dos casos, poderá ocorrer refluxo vesicoureteral (RVU) neste rim único8. A longo prazo, poderão apresentar hipertensão (16%), microalbuminúria (21%) e insuficiência renal (10%)8.
O risco de recorrência varia entre 3-6%, podendo atingir os 8% quando associada a múltiplas anomalias congénitas17,18. Os progenitores deverão realizar também avaliação ecográfica. Se algum for afetado, o risco de recorrência sobe para 15-20%5.
Rim duplex
Caracteriza-se por um rim com dois sistemas pielocaliciais e artérias renais e duplicação parcial ou completa dos ureteres. Na duplicação parcial, mais comum, existe um único ureter ou dois ureteres que se unem proximalmente à inserção na bexiga. Na completa, existem 2 ureteres distintos. O ureter do polo superior insere-se habitualmente inferomedialmente à sua normal inserção, podendo também terminar na vagina ou uretra. O ureter do polo inferior drena lateralmente no trígono vesical. Esta inserção ectópica predispõe a obstrução ou a RVU19.
A prevalência descrita varia entre 1:70-1:50019. É mais frequente à esquerda e unilateral em 83-90% dos casos20,21,22,23,24.
Existem achados ecográficos característicos, mas não patognomónicos: presença de hidronefrose, mais comummente do polo superior do rim; ureterocelo (dilatação quística da porção intravesical do ureter causada pelo estreitamento do orifício ureteral) e megaureter, devido a refluxo ou obstrução da junção vesicoureteral25.
A incidência de aneuplodias cromossómicas não se encontra aumentada25.
A vigilância destas grávidas deve incluir ecografias mensais para avaliação do grau de dilatação e VLA26.
O prognóstico é favorável, com a maioria sem compromisso da função renal. Contudo, apresentam RVU em 70% dos casos e risco aumentado de infeção (20 vezes)25,27.
Anomalias de posição/fusão
Durante a embriogénese, os rins migram da sua posição inicial na pelve para a fossa renal e rodam de uma posição horizontal para vertical28. Qualquer disrupção na migração embriológica normal pode resultar numa anomalia da posição/fusão do rim, das quais se destacam o rim pélvico e o rim em ferradura.
Rim pélvico
A sua prevalência varia entre 1:700-1:1000 nascimentos5. Predomina no lado esquerdo e sexo masculino. É bilateral em 12% dos casos.
Ecograficamente, visualiza-se o rim na pelve acima da bexiga, encontrando-se a glândula suprarrenal a preencher a fossa renal vazia, com bexiga e VLA normais. O Doppler a cores é útil para localizar o hilo do rim ectópico5. Deverá ser realizado diagnóstico diferencial com AR ou ectopia renal cruzada (rins presentes, mas fundidos de um dos lados)5.
Pode associar-se a malformações esqueléticas, cardíacas ou genitourinárias28-30, sendo apenas nestes casos com incidência aumentada de alterações cromossómicas ou genéticas, justificado o cariótipo/array com hibridação genómica comparativa (array-CGH)5,30.
A vigilância destas situações inclui ecografias mensais para diagnóstico de hidronefrose de aparecimento tardio. Adicionalmente, deverá ser proposta ecografia aos progenitores5.
O rim ectópico poderá apresentar diminuição da sua função e associar-se a RVU em 5-30% dos casos e obstrução da junção pieloureteral (OJPU)5,30. O prognóstico é mesmo assim favorável, desde que o rim contralateral desenvolva hipertrofia compensatória30.
Nos casos isolados, não apresenta risco de recorrência30.
Rim em ferradura
A sua prevalência é de 1:400 nascimentos, predominando no sexo masculino5.
Ecograficamente, observa-se fusão dos polos inferiores dos rins, anteriormente aos grandes vasos. Nos planos coronal ou axial, visualiza-se uma ponte de tecido a conectar os rins anteriormente. A bexiga e VLA são normais.
Pode associar-se a aneuploidias cromossómicas (Síndrome de Turner em 30% dos casos ou Trissomia 18 em 20%); a síndromes congénitos (15% dos casos, mais frequente o síndrome de regressão caudal) e ainda, em 1/3 dos casos, a malformações genitais, cardíacas, esqueléticas e do sistema nervoso central (SNC)5. Se isolado ou parte do síndrome de Turner, sem risco de recorrência. No caso da trissomia 18 livre, o risco de recorrência é baixo (1%).
Deverá ser pedido cariótipo/array-CGH fetal e a vigilância incluir ecografias seriadas mensais para pesquisa de hidronefrose de aparecimento tardio.
A maioria dos pacientes é assintomática, embora o risco de infeções, nefrolitíase (70%), hidronefrose (80%) e tumores renais esteja aumentado2.
Anomalias do parênquima renal
Doença Quística Renal (DQR)
A DQR manifesta-se ecograficamente por aumento da ecogenicidade renal. Os rins são considerados hiperecogénicos quando, a partir das 15-17 semanas, apresentam maior ecogenicidade do que o fígado ou o baço (figura 3)5,31.
A avaliação ecográfica de fetos com rins hiperecogénicos deverá incluir: avaliação do VLA, tamanho/volume do rim, diferenciação corticomedular e localização/tamanho dos quistos renais. Adicionalmente, deverá ser questionada história familiar de DQR. A RM poderá ser útil para caracterização de malformações adicionais, sobretudo na presença de oligo/anidrâmnios32.
Na DRQ bilateral, oferecer sempre estudo genético, independentemente da presença de oligoâmnios ou malformações extrarrenais. Na unilateral apenas na presença de malformações extrarrenais32. O estudo genético poderá incluir análise molecular (PKD1,PKD2, PKHD1), array-CGH ou painéis de sequenciação de nova geração33.
O parto deverá ocorrer sempre em unidade com cuidados perinatais diferenciados32.
O diagnóstico diferencial de rins hiperecogénicos é realizado de acordo com o fluxograma 1.
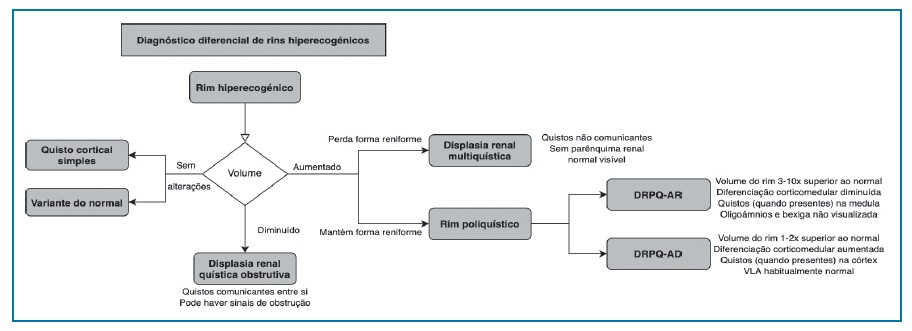
Fluxograma 1 Diagnóstico diferencial de rins hiperecogénicos. Fluxograma adaptado de Paladini (2014). Legenda: DRPQ-AR - doença renal poliquística autossómica recessiva; DRPQ - doença renal poliquística autossómica dominante; VLA - volume de líquido amniótico
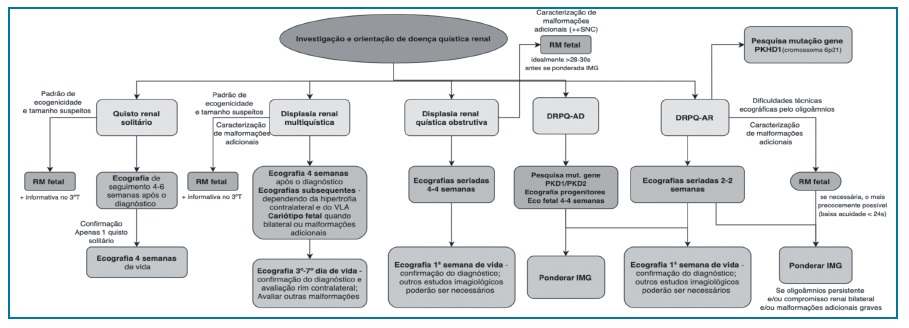
A investigação e orientação da DQR encontra-se descrita no fluxograma 2.
Displasia Renal Multiquística (DRMQ)
A sua prevalência é de 1:1000-5000 nascimentos5,14. Em 75-80% dos casos é unilateral e mais frequente no sexo masculino34,35.
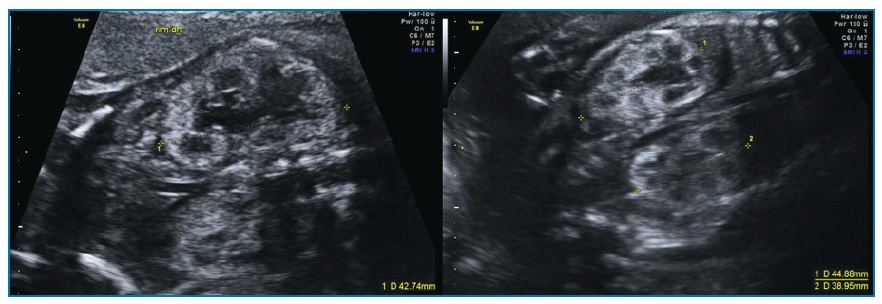
O parênquima renal é substituído por múltiplos quistos irregulares de tamanho variável não comunicantes entre si (figura 4). Não se visualiza tecido nem pelve renal normais. O ureter e a artéria renal ipsilateral podem estar ausentes. Quando bilateral, associa-se a anidrâmnios e ausência de bexiga.
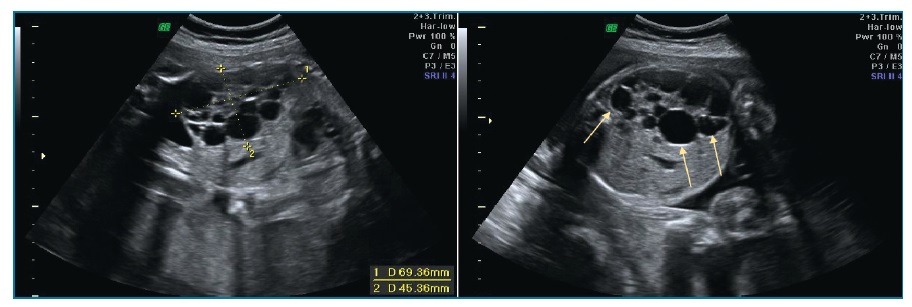
Figura 4 Displasia renal multiquística. Múltiplos quistos de tamanhos variáveis não comunicantes entre si (seta)
Em 25-40% dos casos, pode associar-se a anomalias do rim contralateral ou a RVU, AR e OJPU34,36. Estão também descritas malformações cardíacas, SNC, gastrointestinais ou dos membros. O risco de cromossomopatias é baixo, quando isolada (2-4%), podendo atingir os 15-18% se bilateral ou 25-28% quando associado a outras anomalias 5. Em cerca de 10% dos casos, existe um síndrome associado, sendo os mais frequentes os síndromes branquio-oto-renal, polidactilia-costela curta, Meckel-Gruber e associação VACTERL5. Na forma isolada, o risco de recorrência é de cerca de 3%. Quando associado a síndromes, o risco depende do síndrome em questão5.
As grávidas com DRMQ deverão realizar ecografias mensais para detetar hidronefrose de início tardio. Quando unilateral e em formas isoladas, o prognóstico é bom, sendo o maior fator prognóstico o tamanho do rim contralateral e a presença/ausência de malformações adicionais37. Nas bilaterais, há risco de hipoplasia pulmonar. No período pós-natal, o rim pode sofrer involução progressiva, podendo mesmo desaparecer em aproximadamente 40% dos casos38,39.
Doença Renal Poliquística Autossómica Recessiva (DRPQ-AR)
A DRPQ-AR representa uma forma grave de DQR que envolve o rim e o trato biliar1. É subdividida nos tipos perinatal (40% dos casos), neonatal, infantil e juvenil, de acordo com a idade de aparecimento. A sua prevalência é de 1:20.000 nascimentos. Resulta de uma mutação no gene PKHD11.
Ecograficamente, caracteriza-se por rins aumentados de volume bilateralmente (3-10 vezes o normal) e hiperecogénicos, com fraca diferenciação corticomedular e aparecimento gradual de oligoâmnios a partir do 2º trimestre1. A bexiga habitualmente não é visualizada.
O prognóstico é muito reservado quando a manifestação é perinatal (40-50% morrem no período neonatal precoce por hipoplasia pulmonar). A maioria dos sobreviventes desenvolve hipertensão e ocorre progressão para doença renal crónica com necessidade de transplante renal em 50% dos casos5. Os tipos infantil e juvenil resultam em falência renal crónica, fibrose hepática e hipertensão portal.
Nos casos em que se prossiga com a gravidez, a vigilância ecográfica deverá ser realizada a cada 2-4 semanas com avaliação do tamanho renal, crescimento fetal e VLA1.
O risco de recorrência é de 25%1. Dado o seu padrão AR é fundamental uma consulta de genética para programação de gravidez futura.
Doença Renal Poliquística Autossómica Dominante (DRPQ-AD)
A DRPQ-AD é mais prevalente (1:1000). Raramente diagnosticada no período pré-natal (2-5%), a clínica surge tipicamente entre a 3ª-5ª décadas de vida5. Resulta de uma mutação nos genes PKD1 (85%) ou PKD2 (10-12%)1,5.
Quando a manifestação é pré-natal, os rins encontram-se aumentados de tamanho bilateralmente (1-2 vezes o normal) e hiperecogénicos. A presença de quistos é variável e só raramente cursa com oligo/anidrâmnios1.
A manifestação da doença no período pré-natal é um sinal de mau prognóstico, podendo associar-se a hipertensão durante o primeiro ano de vida. Se oligoâmnios ou hipoplasia pulmonar, a mortalidade a 1 ano poderá atingir os 40% e os 80%, respetivamente1. O risco de recorrência é de 50%, exceto se mutação de novo.
O diagnóstico diferencial entre DRPQ-AD e DRPQ-AR é realizado através de diagnóstico molecular ou história familiar, dada a variabilidade do fenótipo e da altura de apresentação. É importante ter ainda em consideração que rins hiperecogénicos podem ser uma variante do normal, especialmente quando isolados e com VLA normal. Adicionalmente, também poderão surgir associados a outras malformações nos síndromes de Meckel-Gruber, Beckwith-Wiedemann, Bardet-Biedl, trissomia 13 e nefronoftise1.
Dilatações Do Sistema Coletor (DSC)
Um dos achados mais frequentes de diagnóstico pré-natal (1-3% das gravidezes)40. Apresenta um elevado espetro de manifestações, desde dilatações fisiológicas transitórias até uropatias com evolução para doença renal significativa41 (Tabela I).
Tabela I Etiologia renal e extrarrenal das dilatações do sistema coletor renal. Adaptada de Woodward (2002) e Nguyen (2010)42,44
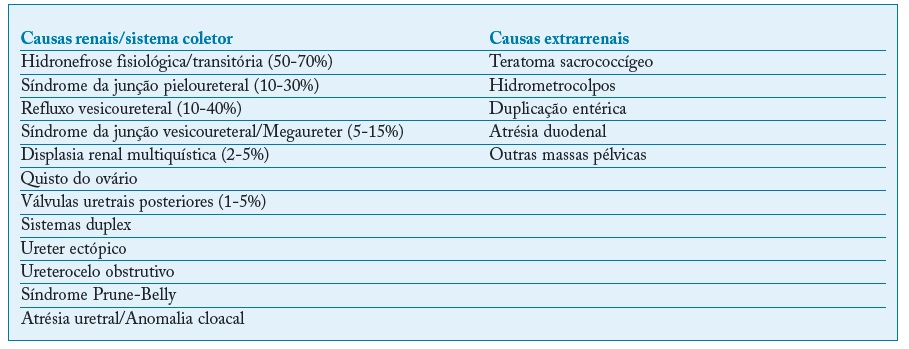
Na avaliação ecográfica, é fundamental avaliar: localização dos rins; quantificação e caracterização da dilatação piélica e/ou calicial; definição da ecogenicidade renal e diferenciação corticomedular; verificação da existência de megaureter; verificação da existência e tipo de quistos; quantificação do VLA; visualização da bexiga (parede, forma, esvaziamento vesical); pesquisa de malformações extrarrenais e sexo fetal.
Para efeitos deste protocolo, as DSC serão apresentadas de acordo com o achado ecográfico (hidronefrose, megaureter e megabexiga).
Hidronefrose
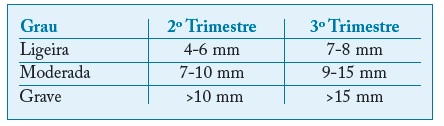
Considera-se hidronefrose a dilatação da pelve renal, com ou sem dilatação dos cálices. Existem vários critérios para o seu diagnóstico, nomeadamente o diâmetro anteroposterior da pelve renal (DPR)42, o sistema de estadiamento da Sociedade de Urologia Fetal43 e o sistema multidisciplinar de classificação da dilatação do trato urinário44,45. O sistema habitualmente preferido pelos obstetras é o do DPR46. Apesar de não haver consenso nos valores que definem uma dilatação clinicamente significativa, classicamente considera-se um DPR superior a 4 mm no 2º trimestre e a 7 mm no 3º trimestre45.
A sua prevalência varia entre 0,6-5,4%, dependendo do critério de diagnóstico utilizado47. É mais frequente no sexo masculino48 e na maioria dos casos, unilateral49.
Ecograficamente, deverá proceder-se à medição do maior DPR obtido em corte transversal do abdómen, com a bexiga fetal vazia (figuras 5 e 6). De acordo com a medição obtida em cada trimestre é determinado o grau de dilatação piélica (Tabela II).
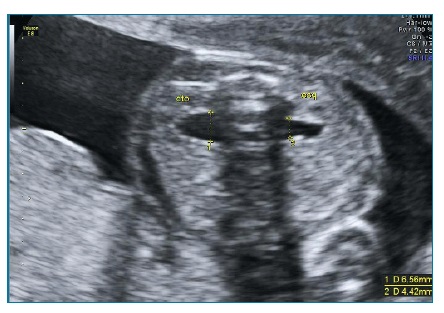
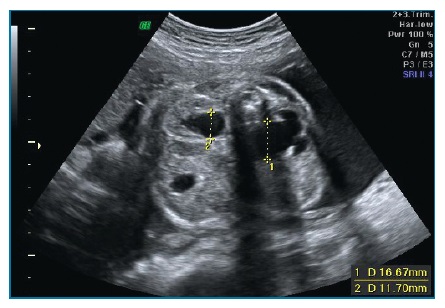
Figura 6 Hidronefrose grave à esquerda (DPR 16,67 mm) e moderada à direita (DPR 11,7 mm) em ecografia do 3º trimestre da gravidez
Tabela II Classificação da gravidade da hidronefrose de acordo com o trimestre de gravidez. Adaptado de Nguyen (2010)44
A etiologia mais frequente é a hidronefrose fisiológica/transitória, que habitualmente resolve durante o terceiro trimestre ou após o parto, não representando doença significativa50,51. A presença de dilatação pielocalicial sem dilatação do ureter e da bexiga, mais comummente unilateral, é altamente sugestiva de OJPU36,44. Já se a causa for o RVU, será visível dilatação intermitente da pelve renal, de grau variável, que aumenta após a micção36.
Nas formas isoladas, existe baixa associação a alterações cromossómicas. Pode-se associar a malformações no rim contralateral, como DRMQ, ectopia ou AR. No contexto sindrómico, associa-se a malformações cardíacas, displasias esqueléticas e anomalias da coluna vertebral, justificando nestes casos cariótipo fetal e/ou array-CGH.
O seguimento ecográfico é dependente da idade gestacional ao diagnóstico, da gravidade da dilatação, se é bilateral ou se há suspeita de obstrução do trato urinário inferior45. A orientação específica encontra-se descrita no fluxograma 3. Não existe evidência para antecipação do parto.
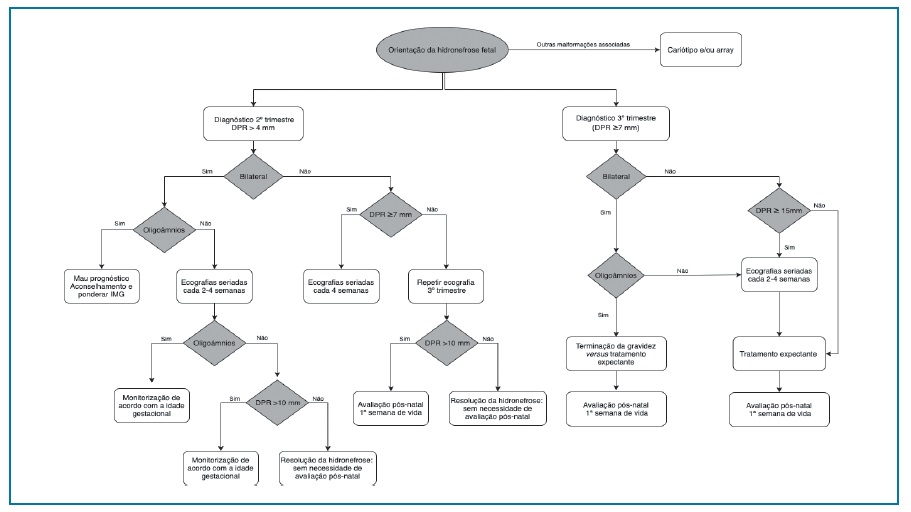
Fluxograma 3 Orientação da hidronefrose fetal. Legenda: DPR - diâmetro da pelve renal. IMG - interrupção médica da gravidez. Fluxograma adaptado de Liu41 (2014).
Até 88% das DSC resolvem no período pré-natal ou neonatal precoce52. Em cerca de 20% dos casos, poderá requerer seguimento pós-natal e eventualmente cirurgia42. A hidronefrose moderada é geralmente progressiva e em mais de 50% dos casos a cirurgia é necessária nos primeiros 2 anos de vida.
Megaureter
Hidronefrose associada a dilatação do ureter, sem dilatação da bexiga (figura 7)44. Quando unilateral, bexiga e VLA são normais. A sua prevalência é de 1:6500 nascimentos.
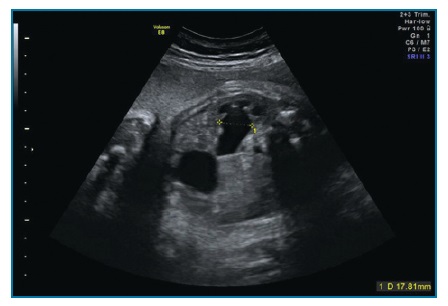
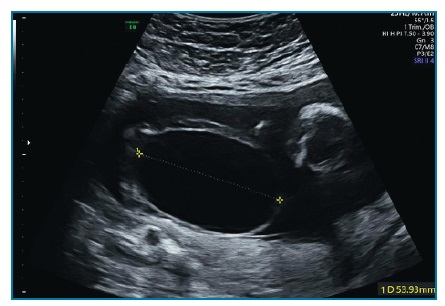
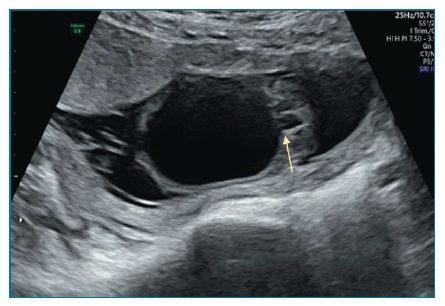
Figura 9 Dilatação da bexiga por válvulas uretrais posteriores (sinal em buraco de fechadura - seta)
Pode ser primário, quando resulta de uma anomalia funcional ou estrutural da JVU, ou secundário a uma anomalia da bexiga ou uretra2. As causas mais frequentes são a obstrução da junção vesicoureteral (OJVU), o RVU, ureterocelo e ureter ectópico5. Pode associar-se a rim duplex, RVU e anomalias genitais, sobretudo em fetos do sexo feminino.
A vigilância deverá incluir ecografias mensais.
Megabexiga
No 1º trimestre define-se como diâmetro longitudinal da bexiga superior ou igual a 7 mm (figura 8)1. A partir do 2º trimestre, as definições não são tão claras: diâmetro longitudinal da bexiga > (idade gestacional em semanas + 2 mm); espessura da parede da bexiga >3 mm ou bexiga aumentada de tamanho (2º ou 3º trimestre) sem se visualizar esvaziamento ao fim de 45 minutos1,53.
A sua prevalência ronda 1:3000 nascimentos. As causas podem ser obstrutivas (válvulas uretrais posteriores (VUR), atrésia uretral ou disgenesia cloacal) ou não obstrutivas (doenças neurológicas, cromossómicas ou genéticas, das quais se destaca o síndrome de megabexiga-microcolón-hipoperistaltismo intestinal (MMHI))1,5.
Ecograficamente, nas VUR (25% dos casos, apenas no sexo masculino) verifica-se dilatação da uretra proximal decorrente da obstrução incompleta ou intermitente da uretra (sinal do ‘buraco de fechadura’ - figura 9), dilatação e hipertrofia da bexiga (espessura da parede > 3mm) e graus variáveis de hidroureteres, hidronefrose, displasia renal cística, oligoâmnios (>17 semanas) e hipoplasia pulmonar36.
No caso de atrésia uretral (condição mais grave) não há excreção de urina, visualizando-se a bexiga muito aumentada precocemente (12 semanas). A partir das 16/17 semanas associa-se a anidrâmnios. É uma condição letal5.
O síndrome MMHI, mais frequente no sexo feminino, cursa com hidronefrose, megabexiga, hidrâmnios e dilatação do estômago1, sendo o estudo genético útil para diagnóstico.
Em aproximadamente 8-20% dos casos encontram-se alterações cromossómicas (trissomia 13, 18 ou 21) e em 40% dos casos malformações cardíacas ou gastrointestinais, sendo imperativa uma ecografia detalhada e cariótipo/array-CGH.
Dependendo da gravidade e da idade gestacional, pode ser formulada IMG, transferência para centro diferenciado para derivação vesical, terminação da gravidez ou atitude expectante com cirurgia pós-parto.
A intervenção fetal (shunts vesicoamnióticos e cistoscopia fetal) para causas obstrutivas permanece controversa, uma vez que se associa a elevadas taxas de complicações, sem evidência de melhoria da sobrevida ou da função renal a longo prazo45. Testes de avaliação da função renal, como a concentração de b2-microglobulina ou cistatina-C (marcadores da função glomerular) na urina fetal ou dos eletrólitos e osmolalidade urinária (marcadores da função tubular fetal), obtidos por vesicocentese36,41, parecem não ser suficientemente fidedignos para predizer a função renal pós-natal54.
Se for decidido prosseguir com a gravidez realizar ecografias mensais para avaliar a sua evolução e o VLA.
Em alguns casos, sobretudo para dilatações entre 8-12 mm, há resolução espontânea. Diâmetros superiores a 17 mm são mais frequentemente de causa obstrutiva, com progressão para uropatia grave, verificando-se elevada mortalidade perinatal nos casos mais graves12. Nos que sobrevivem, cerca de 30% necessitam de diálise e/ou transplante renal antes dos 5 anos de idade.
Contribuição individual de cada autor
Mariana Dória - Pesquisa bibliográfica, processamento da recolha de dados, revisão científica sobre o tema, escrita do manuscrito e revisão final do trabalho
Ana Mesquita Varejão - Apoio na pesquisa bibliográfica e processamento da recolha de dados e revisão final do trabalho
Inês Sarmento Gonçalves - Revisão final do trabalho
Fátima Soares - Revisão final do trabalho

