











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Stickler syndrome (SS) is a group of hereditary heterogeneous conditions affecting connective tissue with an estimated prevalence of 1 in 7.500 to 9.000 newborns1-3 and it is the leading cause of rhegmatogenous retinal detachment in childhood4.
Both autosomal dominant and recessive forms have been described, all resulting from mutations in several genes encoding collagens type II, IX and XI, which are expressed within the vitreous, skeleton, and inner ear1,5-7. Based on its underlying genetic collagen defect, SS is subdivided into several subtypes. The features most commonly associated includes ocular abnormalities (high myopia, retinal detachment, vitreous anomalies, cataracts and/or glaucoma) 5,6, sensorineural hearing loss2,5,6,8, craniofacial dysmorphisms (flattened facial profile known as midface hypoplasia, malar hypoplasia, flattened or broad nasal bridge, anteverted nares and, in some cases, micro/retrognathia as part of a Pierre-Robin-sequence-associated cleft palate) 3,5-8,9 and skeletal manifestations (joint hypermobility, skeletal dysplasia and progressive arthropathy)5,6,8. There are limited genotype-phenotype correlation and SS shows significant inter and intrafamilial phenotypic variability3,6,7.
The diagnosis of SS is clinically based. At present, no consensus minimal clinical diagnostic criteria exist3. Pathogenic variants in one of six genes (COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, COL9A3) have been associated with SS; because a few families with features of SS are not linked to any of these six loci, pathogenic variants in other genes may also cause the disorder3.
Type 1 SS is the most common, accounting for approximately 80-90% of the cases1,6. It has autosomal dominant transmission. and is caused by heterozygous pathogenic variants in COL2A1 gene which encodes type II procollagen1,6. A smaller proportion of the autosomal dominant form is caused by heterozygous pathogenic variants in COL11A1, COL11A2 and BMP4 genes. Type 2 SS and Type 3 SS (formerly known as Non-Ocular SS) are caused by heterozygous pathogenic variants in COL11A1 (which encodes type XI procollagen) and COL11A2 (which encodes type XI procollagen), respectively10,11. Type 7 SS (the only non-collagenous form) is caused by mutations in BMP4 gene1. Autosomal recessive transmission has been described in some families, with mutations in genes encoding type IX collagen: COL9A1 -Type 4 SS12, COL9A2 - Type 5 SS13 and in COL9A3 -Type 6 SS4. Mutations in LOXL3, an enzyme that crosslinks type II collagen has been described in Type 8 SS14.
Current literature lacks a detailed prenatal phenotype of SS and so prenatal diagnosis is rare. Here we describe the prenatal appearance and ultrasound reports of a type 2 SS case without known familial history.
Case report
A 33-year-old, healthy woman was referred to perform the routine antenatal ultrasounds. Her husband had severe myopia since an early age and the couple was non-consanguineous. They have a healthy 6-year-old son. No relevant familial history was reported.
The first screening ultrasound, at 11+5 weeks’ gestation, was normal. The second trimester ultrasound, performed at 22+5 weeks, revealed increased nuchal fold (8.2 mm), micrognathia, macrophtalmia, facial edema and bilateral mild hydronephrosis (6 mm), (Figure 1 and 2). The amniocenteses (with aneuploidy screen and array comparative genomic hybridization study) performed at 23+1 weeks’ gestation was normal. Fetal echocardiogram, at 25+2 weeks revealed no abnormalities. A fetal cerebral Magnetic Resonance Imaging performed at 24+5 weeks’ gestation only showed nuchal/craniofacial edema.

Figure 1 and 2. 2D- Ultrasound at 22+5 weeks’ gestation revealing increased nuchal fold, micrognathia and prominent lips.
Subsequent ultrasounds, at 29+1 weeks and 34+5 weeks’ gestation, revealed persistent craniofacial edema, micrognathia, flat midface and prominent eyeballs and lips (Figure 3, 4 and 5). Based on these facial alterations SS suspicion was raised after a multidisciplinary discussion.
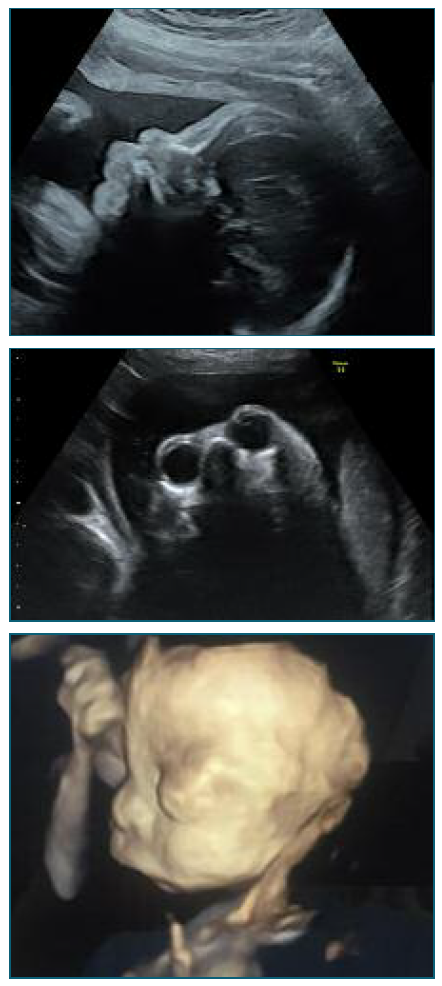
Figure 3, 4 and 5. 2D Ultrasound at 29+1 weeks’ gestation and 2D and 3D Ultrasound at 34+5 weeks’ gestation demonstrating micrognathia, and prominent eyeballs and lips.
The pregnancy was otherwise uneventful and at 40+6 weeks’ gestation a 3705 g boy was born by cesarean section with an Apgar score of 9/10/9 at 1’, 5’ and 10’ minutes respectively. At 10 minutes of life, the newborn started hypoxemia and was admitted to the Neonatal Intensive Care Unit. Postnatal examination revealed axial hypotonia, craniofacial dysmorphia with flat face and prominent forehead, bilateral exophthalmos, glossoptosis, retrognathism, wide rhomboid fontanelles, low-set ears, and posterior cleft palate (Figure 6 and 7). 3D cerebral computed tomography was performed and revealed mandibular hypoplasia, large sagittal suture, and large anterior fontanelle. The oph-thalmologic examination showed marked choroiditis of the retina and near-sightedness and he failed the universal neonatal hearing screening.
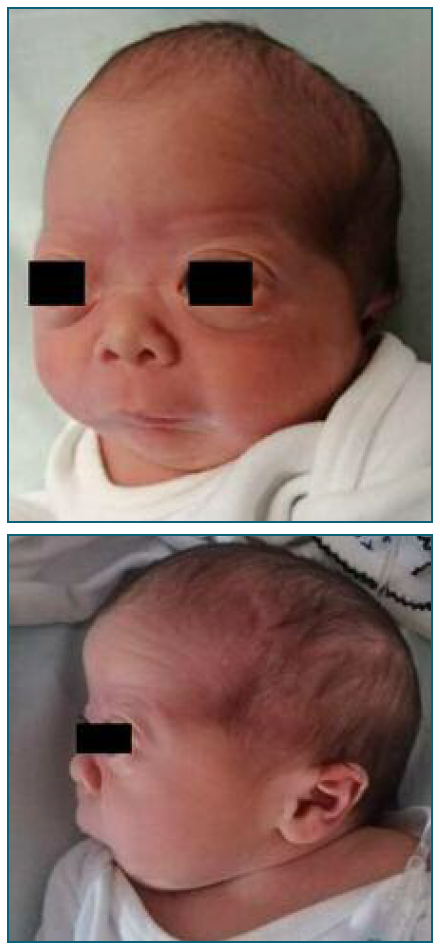
Figure 6 and 7. Postnatal facial abnormalities typical in Stickler syndrome: craniofacial dysmorphia with flat face and prominent forehead, bilateral exophthalmos, retrognathism, wide rhomboid fontanelles, low-set ears (frontal and lateral view).
Genetic studies found a deletion of the COL11A1 gene, variant c.2952+1G>T confirming SS type 2. After the diagnosis, it was recommended to the newborn’s parents to perform carrier testing for the familial variant, although it wasn’t performed yet, by their choice.
Discussion/Conclusion
SS of a group of hereditary collagenopathies characterized by increased risk of rhegmatogenous retinal detachment and developmental abnormalities in vitreous and other tissues containing cartilage. The diagnosis remains difficult to confirm prenatally unless family history and/or morphological examination of the parents directs to a familial form. In this case the newborn father presented some facial characteristics (exophthalmos and prominent forehead) that in combination with the history of severe myopia let the multidisciplinary team raise the suspicion of SS based on ultrasound features (retrognathism, flat midface, prominent eyeballs, and lips). In the newborn the diagnosis is often missed and diagnosed mostly as Pierre Robin sequence (PRS). Approximately half of all individuals with PRS have an underlying syndrome, of which SS is the most common3,15. The PRS, which includes micrognathia, glossoptosis, upper airway obstruction and frequently cleft palate, is observed in approximately 30% of SS cases4,7,15. In SS cases that result from collagen II mutation, facial features can become less prominent in adulthood, whereas those with collagen XI mutations, like the one present, these alterations usually persist but can become milder4,7. Cleft palate is described in approximately 45% of Type-1 SS and 28% of Type-2 SS patients14. In this case, the newborn had a posterior cleft palate that was missed during prenatal ultrasound screening. Prenatal detection rates of isolated cleft palate are low and remain challenging. In the retrospective study of Offerdal et al. 16 based on 49.314 deliveries, none of the 24 clefts palate not associated with a cleft lip had been prenatally diagnosed. Of these, 42% were strictly isolated, while 58% were associated with other abnormalities: chromosomal (17%), syndromic (38%) or morphological (4%)16. PRS is relatively rare, but the mortality rate has been reported to be as high as 30%17. The most concerning features are micrognathia and glossoptosis because the combination of these two can obstruct the airway causing respiratory distress and poor oral feeding. In this case, at 10 minutes of life, the newborn developed respiratory difficulties and needed to be admitted in the Neonatal Intensive Care Unit and showed some feeding difficulties on the first days of life. Thus, prenatal suspicion of PRS could be important to the neonatology team that will assist birth for a possible airway emergency.
Mutations in COL11A1 gene have been associated with autosomal dominant disorders such as Type-2 SS, Marshall Syndrome, and Fibrochondrogenesis 1 a recessive skeletal dysplasia18. A clinical overlap is commonly observed between Marshall Syndrome and SS cases. It is unclear whether they are distinct entities or different clinical manifestations of a single syndrome. Even expecting a more severe phenotype in a patient affected by Marshall syndrome, striking ocular hypertelorism and ectodermal abnormalities, when facial characteristics are not very evident the diagnosis can be difficult to define18,19.
The pathognomonic hallmark of SS is the congenital abnormality of vitreous embryogenesis (the only exception being type-3 SS). In fact, SS is frequently diagnosed by ophthalmologists due to the high rate of ocular complications. Affected individuals are at significantly increased risk for retinal detachment and blindness. Early diagnosis is critical to improving visual outcomes in these patients. Risk of retinal detachment in individuals with type-2 SS has been less studied than in those with type-1 SS but has been reported to be approximately 40%8. At birth, newborn ocular evaluation showed near-sightedness. Nowadays, at age of 3 he has severe myopia and was recently submitted to cataract surgery.
Hearing loss is a common feature of SS. A meta-analysis including 313 patients in 46 studies, revealed that 63% of patients with type-1 SS had hearing loss, compared with 82.5% in type-2 SS, and in this last group, hearing loss was more severe2,5. At birth, the newborn failed the universal neonatal hearing screening and nowadays the patient auditory evoked potentials and the auditory steady-state response suggest mild to moderate hearing loss.
The diagnosis of SS is based on clinical features but there are no validated consensus nor diagnostic criteria to define it3, which turns difficult the diagnoses unless a high level of suspicion exists.
This case illustrates that prenatal ultrasound findings may provide precious clues for early recognition of SS. Without known familial history, prenatal diagnosis is very difficult, but early recognition of this rare entity would be useful for counselling these parents allowing them to make informed choices regarding further management of their affected children. Additionally, it would also prove essential to prepare neonatal team assistance at time of birth and optimize the surveillance of these patients.
Authors’ contributions
AC Borges: Substantial contributions to the conception or design of the work; to the acquisition, and interpretation of data for the work; manuscript writing; Final approval of the version to be published.
C Marques: contributions to the acquisition, and interpretation of data for the work; Review of the manuscript; Final approval of the version to be published.
N Teixeira: contributions to the acquisition, and interpretation of data for the work; Review of the manuscript; Final approval of the version to be published.
A Gonçalves-Rocha: contributions to the interpretation of data for the work; Review of the manuscript; final approval of the manuscript.
A Cadilhe: contributions to the interpretation of data for the work; Review of the manuscript; final approval of the manuscript.
Statement of Ethics
Ethics approval: This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the local Ethics Committee, approval number [6_2023].

