











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
INTRODUCTION
Dyskeratosis congenita (DC) is a rare genetic disease that affects more males and presents with typical cutaneous and systemic signs, such as the mucocutaneous triad (dyspigmentation, nail dystrophy, leukoplakia). We present a case in a female patient, who already presented the typical signs at 2 months of age (below the median age reported in the literature - 14 years of age)1 as well as atypical patterns of progression of onychodystrophy and adermatoglyphia and positive result for short telomeres.
CASE REPORT
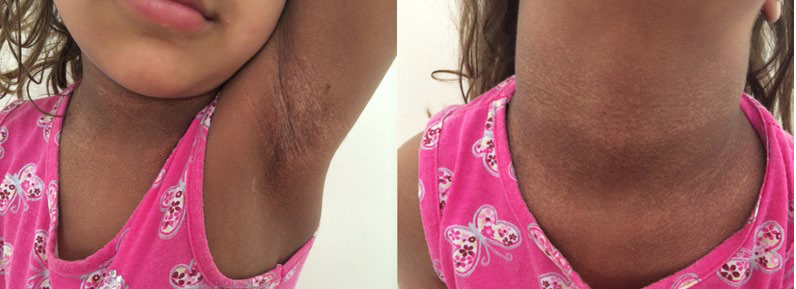
A 2-years-old female child, the second daughter of non-consanguineous parents, born by cesarean delivery after na uneventful pregnancy, was observed with reticulated hyperpigmentation and hypochromic areas in the face (nasal dorsum, upper lip and lower eyelids), cervical region, armpits, inguinal region, hands (back and palm) and dorsum of the feet. She also had nail dystrophy in the toes, not in the fingers, but here we could observe adermatoglyphia affecting all the fingers.
There was also a 2 x 1.5 cm plaque of oral leukoplakia in the papillary tongue (Fig.s 1, 2 and 3). These skin lesions had started at the age of two months. She presented also a minor speech delay, but no alterations in other development parameters.
There are no other complaints and the parents denied similar cases in the family. Biochemical tests were normal. The investigation of the telomeric length of leukocytes in peripheral blood, carried out in the laboratory of the Hospital das Clínicas, Faculdade de Medicina de Ribeirão Preto, São Paulo state, Brazil, showed a telomer length below the expected range for the patient´s age (short telomere), corroborating the diagnosis of dyskeratosis congenita. Currently, the patient is six years old and has developed no further complications during the multidisciplinary follow-up and there was no progression in the area of leukoplakia or dyspigmentation. She does not show any discomfort or social damage regarding the skin manifestations.
The parents are well informed and involved in the care of their daughter but they chose not to perform genetic evaluation.
DISCUSSION
DC was first described in 1906 by Zinsser, and then characterized as a clinical entity in 1926 by Engman and Cole, thus it became known as the Zinsser-Cole-Engman syndrome.2,3 Since 1998, at least eleven genes associated with DC have been recognized, with nine of them related to maintenance of the length of the telomeres. The inheritance patterns of DC are X-linked-recessive (XLR) for the DKC1 gene, autosomal dominant (AD) for TERC, TERT, TINF2, RTEL1 and ACD and autosomal recessive (AR) for TERT, NOP10, NHP2, WRAP53, CTC1, USB1, RTEL1 and ACD.2-4The most common mutation is in DKC1, which explains, in part, the trend towards males (approximate ratio of 3.2:1).2,6,7However, about 30% to 40% of patients may not have an identifiable mutation in the genetic evaluation.5 The clinical features of DC are highly variable, even within families with the same mutation, with variable penetrance and phenotypic differences. The estimated annual incidence is 1 in 1 million individuals.2,6,7
The classic clinical presentation of DC starts at around five to twelve years of age, characterized by the mucocutaneous triad: abnormal skin pigmentation, nail dystrophy and oral leukoplakia. Shimamura et al (2010) evaluated 550 reports published from 1910 to 2009. The age at diagnosis range from 0 to 75 years, but was mostly between 0 to 30 years, with median age of onset at 14 years.1 Skin changes occur in approximately 90% of patients as reticulated hyperpigmentation in different body regions. Nail dystrophy is present in about 90% of patients, with a tendency to involve initially the fingernails and later toenails.4 Other skin changes include cutaneous atrophy, hyperhidrosis and hyperkeratosis of the palms and soles, telangiectasia, cracking, fissuring, bullae and loss of dermal ridges.8 Epidermal atrophy, basal cell degeneration and increased melanophages are found in histopathology.9
Oral leukoplakia, the most specific clinical manifestation, affects about 80% of DC patients, and involves the tongue, oral mucosa and oropharynx. Malignant transformation occurs mostly in areas of leukoplakia: squamous cell carcinoma (SCC) occurs in about 30% of individuals within 10 to 30 years.3 Severe periodontal destruction, gingival inflammation, bleeding and bone loss are other significant associations.
The patient in this report has the typical DC triad of leukoplakia, hyperpigmentation in the classic regions and onychodystrophy was significant on the feet, not on the hands, contrasting with the usual DC trend. Adermatoglyphia, another potential finding in DC, was evident on all fingers (Fig.s 1, 2 and 3).

Figure 1 Hands and feet showing bilateral reticulated hyperpigmentation. Onychodystrophy is evident in the feet, being more severe in the right foot. Bilateral adermatoglyphia is also evident on the hands.

Bone marrow failure (BMF) is the leading cause of death in 60% to 80% of patients.2,3It usually starts before the age of 20, and approximately 90% of patients have this symptom by the age of 30.10 The progressive shortening of telomeres with repeated cell divisions reaches its critical threshold and, thereafter, cell proliferative capacity becomes limited. Tissues with a high proliferative rate, such as hematopoietic, mucosa and epithelium, are mainly affected.1 In about 90% of patients BMF initially presents as gradual thrombocytopenia, before developing peripheral cytopenia in other hematological lineages.3 Hematological changes were not evident in this patient, as would be expected at this age, but periodic hematological follow-up is mandatory for early identification of BMF.
Malignancy and pulmonary disorders are other important complications in the adult with DC. Telomere shortening may be associated with chromosome instability, which promotes tumorigenesis.3The cumulative incidence of malignancy is around 40% to 50% at 50 years of age. The most common tumors are SCC of the skin, tongue, larynx and bronchial tree, in addition to leukemia, with or without previous myelodysplastic syndrome, and adenocarcinoma of the gastrointestinal tract.2,3Pulmonary fibrosis, present in about 80% of adult patients due to the high rate of pulmonary epithelium losses, causes difficulties in gas exchange and abnormalities in pulmonary vascularization and is also an important cause of mortality.3 The patient in this case undergoes periodic dermatological and non-invasive pulmonary monitoring, using techniques such as spirometry, to avoid exposure to x-rays due to the increased risk of neoplasia.
DC may cause multisystemic defects in varying aspects, including: development delay; microcephaly; ophthalmological abnormalities; premature aging; lung disease; hepatic cirrhosis; hypogonadism and malignancy; alopecia; and premature fatigue.7 DC has two clinical variants, Revesz syndrome and Hoyeraal-Hreidarsson syndrome (HHS), differing in severity. The classic symptoms of DC, such as mucocutaneous manifestations and BMF, are common for both syndromes; however, both have serious deficiencies in other systems. Revesz syndrome is associated with intrauterine growth retardation, psychomotor retardation, bilateral exudative retinopathy, cerebral calcification and cerebellar hypoplasia. Hoyeraal-Hreidarsson syndrome is associated with intrauterine growth retardation, severe mental retardation, immunodeficiency and pancytopenia.
The diagnosis of DC is made when at least two major and two minor criteria are present.3 The major criteria are the three components of the mucocutaneous triad and BMF.
The minor criteria include components associated with multisystem disorders: epiphora, development delay, periodontal disease, esophageal stenosis, premature aging or hair losses and development of malignant lesions. Telomere shortening is not specific to DC, but telomere size has a sensitivity of 97% and specificity of 91% when it is below the first percentile in lymphocytes for the patient´s age.3 The patient in this case has three major criteria (onychodystrophy, reticulated hyperpigmentation and oral leukoplakia), one minor criterion (delay in speech development) and an examination showing telomeres below the first percentile for her age. The most relevant differential diagnoses associated with telomere shortening are Shwachman-Diamond syndrome and Fanconi anemia. Fanconi anemia frequently presents with pancytopenia, café au lait, hyper- or hypopigmentation areas, and has a strong association with endocrinological abnormalities. Shwachman-Diamond syndrome often presents with neutropenia and intestinal malabsorption due to pancreatic insufficiency. In both cases short stature is common.1
BMF is treated according to the degree of cytopenia and does not respond adequately to immunosuppressive therapy.2 Hematopoietic stem cell transplantation (HSCT) is the existing curative modality for BMF.11 Androgens such as oxymetholone and danazol may be a therapeutic alternative, as they possibly interfere with erythropoiesis, stimulating hematopoietic stem cells and modulating telomerase.3 Danazol has fewer adverse effects, especially with regard to the masculinization of women.3
The management of dermatological changes involves prevention and symptomatic treatment: photoprotection to reduce the risk of skin malignancy, keratolytic agents, to soften hyperkeratosis, moisturizers to improve dry skin in regions with poikiloderma and cutaneous atrophy.5 The management of nail changes aims to prevent accidental injuries during daily activities, but there is no specific treatment for onychodystrophy in these cases. The patient must reduce excessive exposure to detergents and other irritants, avoiding prolonged use of artificial nails, and must periodically polish them to reduce discomfort and possible accidents. The use of biotin can help to strengthen brittle nails. Good dental hygiene may help prevent early dental loss and delay the development of malignancy of the tongue.5
Laser therapy, such as carbon dioxide laser, is effective in treating oral leukoplakia.2 Surgical excision is curative form for most malignant lesions without metastasis, but radiotherapy and chemotherapy agents are not recommended.1
Therefore, prevention is important by avoiding exposure to potentially carcinogenic substances, such as alcohol, tobacco, and also by vaccination for the human papiloma virus (HPV).5
Genetic testing of the patient and relatives and genetic counseling is important to help the patient and her family to have a more realistic perspective about the disease expectations and limitations. If the patient has a de novo mutation and parents do not have mutations, the chances of having another child with DC are low.5
CONCLUSION
Patients with DC should receive periodic multidisciplinary follow-up to identify and treat early the multisystemic component and potential lifelong complications. This report describes a female patient who developed early mucocutaneous manifestations and telomere length below the first percentile for her age, corroborating the diagnosis of DC, but still no other manifestations by the age of six years old.

