











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A síndrome de Klippel-Feil (SKF) é uma doença que afeta maioritariamente a coluna vertebral, causando fusão de um número variável de vértebras cervicais. Pensava-se tratar de uma doença rara; contudo, de acordo com os últimos estudos, crê-se que a sua prevalência seja de um em cada 172 nascimentos.1 É mais frequente no sexo feminino e manifesta-se através de uma tríade clínica, presente em 40 a 50% dos doentes, caracterizada por pescoço curto, baixa linha de implantação do cabelo e redução da capacidade de mobilização cervical.1-3
Para além dos sintomas mais comuns associados a esta patologia, como cervicalgia, cefaleias e alterações da mobilidade cervical, podem surgir complicações mais graves, destacando-se a estenose medular e défices neurológicos. 4-5
Estão descritos três subtipos de acordo com a porção de vértebras cervicais afetadas: o tipo I apresenta um único segmento de vértebras cervicais fundidas; o tipo II inclui múltiplos blocos não contíguos; já o tipo III comporta múltiplos blocos de vértebras cervicais fundidas de forma contígua. 2
O diagnóstico é mais frequente na infância; contudo, em pacientes assintomáticos, sem manifestações evidentes ao exame objetivo, o diagnóstico pode surgir apenas na idade adulta. 4,6
Associadamente estão descritos defeitos ao nível de outros sistemas, nomeadamente problemas auditivos, alterações renais, crânioencefálicas e cardiovasculares. 1,3,6
A patogénese desta síndrome está associada a falhas na segmentação e diferenciação dos somitos cervicais durante a embriogénese, podendo ser causada por uma mutação esporádica ou associada a vários genes. 3,7-8
O caso que se descreve reúne grande parte das alterações descritas até hoje relativamente a esta síndrome. O utente foi acompanhado ao longo de cerca de quatro anos pelo seu médico de família (MF), que progressivamente foi identificando alterações de vários órgãos e sistemas, culminando no diagnóstico de SKF. Neste caso, a articulação do MF com várias especialidades e com os recursos disponíveis na comunidade permitiu não só o diagnóstico e a gestão clínica deste utente, mas também o seu acompanhamento a nível social, espiritual e relacional.
Este caso clínico tem como objetivo a sensibilização da comunidade médica para esta síndrome rara e para as comorbilidades associadas. Ademais, pretende relatar o papel preponderante do MF no acompanhamento longitudinal deste utente e da sua família ao longo das várias etapas da patologia.
Descrição do caso
Utente, sexo masculino, 15 anos, pertencente a família nuclear no estadio V de Duvall, residente com os pais e irmã de 19 anos. Frequenta o décimo ano de escolaridade com aproveitamento. Sem antecedentes pessoais de relevo, alergias conhecidas ou toma habitual de medicação. Gravidez vigiada, sem identificação de alterações morfológicas nas ecografias realizadas, segundo a mãe.
Recorre à primeira consulta com MF, acompanhado pela mãe, por queixas de dor cervical e torácica agravadas com os movimentos de rotação e flexão da coluna, com cerca de três anos de evolução, intermitente, de ritmo mecânico. Nega outros fatores de agravamento. Refere alívio ligeiro com a toma de analgésico. Nega outros sinais ou sintomas associados. Nega trauma associado.
Ao exame objetivo apresenta implantação baixa da linha do cabelo, pescoço curto, cifose cervical (Figura 1), atitude escoliótica com desvio direito da coluna dorsal (Figura 2) e retificação da coluna lombar. Associadamente apresentava dificuldade na realização de movimento de rotação e inclinação lateral da coluna cervico-dorsal para a direita, sem compromisso da extensão e flexão. Sem alterações da força muscular ou sensibilidade, com reflexos osteo-tendinosos mantidos. Associadamente, à auscultação cardíaca identifica-se click sistólico de ejeção. Sem outras alterações a destacar.

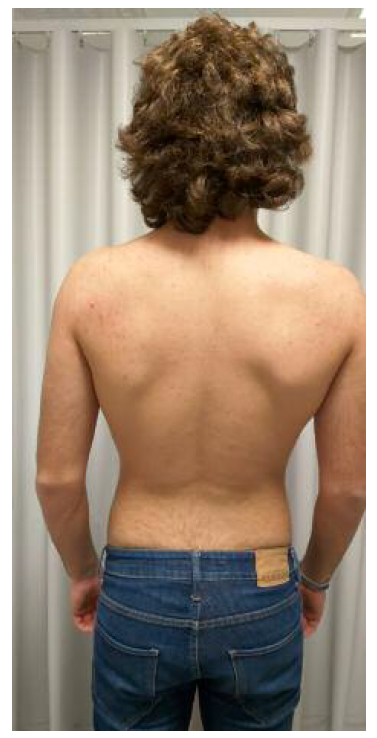
Figura 2 Exame objetivo - inspeção póstero-anterior: atitude escoliótica com desvio direito da coluna dorsal.
Colocando como hipótese diagnóstica mais provável uma malformação da coluna vertebral optou-se por solicitar exame imagiológico, no sentido de identificar alterações da coluna vertebral. Foi também pedida avaliação analítica com o intuito de excluir doenças de envolvimento sistémico, pensando em patologias como a espondilite anquilosante e a artrite juvenil idiopática. Foram solicitados também exames cardíacos, nomeadamente eletrocardiograma de doze derivações e ecocardiograma transtorácico, para investigação da alteração auscultatória.
A tomografia computorizada (TC) da coluna vertebral identifica “retrolistese grau I em IV de C5 sobre C6. Bloco somático em C2-C3, C6-C7, D1-D2-D3-D4-D5, hemivértebra esquerda em D3 e vértebra ‘em borboleta’ em D5. Fusão parcial dos primeiros arcos costais à direita. Deformação em ‘S’ da região alta da ráquis dorsal. Invaginação basilar. Assimilação de C1. Ausência de fusão na linha média dos arcos costais anteriores e posterior de C1. Ligeira deformação cifótica do ráquis cervical. Retificação relativa da coluna lombar por perda da normal curvatura lordótica fisiológica” (Figuras 3, 4 e 5). Associadamente é também descrita, neste relatório, a ausência do rim direito.
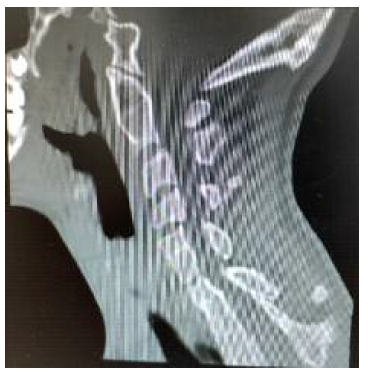

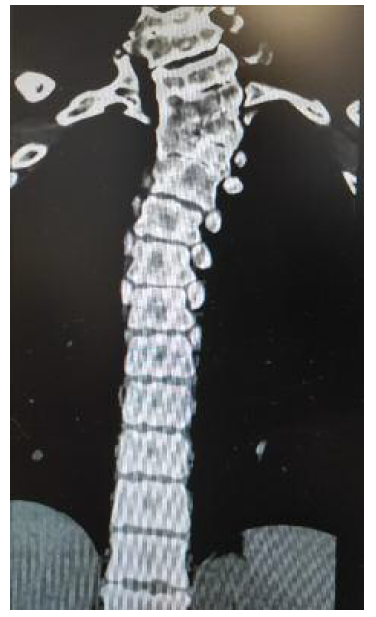
Dos exames cardíacos solicitados destaca-se válvula aórtica bicúspide, sem outros achados anómalos.
Dada a presença de alterações da coluna vertebral associada a duas outras alterações orgânicas (cardíaca e renal) é realizada pesquisa bibliográfica e o SKF surge como a principal hipótese diagnóstica.
O utente é referenciado a consulta de ortopedia, com o intuito de avaliar eventual necessidade de intervenção cirúrgica por parte da especialidade, e a consulta de medicina física e reabilitação (MFR) do hospital de referência, com o objetivo de proporcionar alívio da dor e eventual plano de reabilitação e de reforço muscular. No que se refere à consulta de ortopedia, o MF é informado de que o caso seria discutido em reunião inter-hospitalar da especialidade, ficando a aguardar parecer sobre eventual plano terapêutico.
O utente é também referenciado a consulta de cardiologia pediátrica com exclusão de outras malformações do aparelho cardiovascular.
Solicita-se estudo do aparelho genito-urinário através da ecografia renal e vesical para esclarecimento das alterações descritas previamente. Este exame descreve provável agenesia do rim direito e rim esquerdo vicariante com cerca de 14,4 x 6,2 x 6,1cm, sem outras alterações associadas. Neste sentido, é contactada diretamente a especialidade de nefrologia que, após o enquadramento do caso e dada a ausência de alterações na função renal do utente, refere não existir fundamento para referenciação de momento.
Relativamente à consulta de MFR, é decidido que o utente não beneficia de apoio da especialidade, não apresentando restrições à prática de exercício físico e tendo alta da consulta, com indicação para exercícios de reforço muscular. É realizado um pedido interno de consulta de cirurgia pediátrica, onde é descartada a necessidade de intervenção.
Ao longo da marcha diagnóstica e após pesquisa cuidada das possíveis alterações associadas à SKF optou-se pela avaliação de eventual envolvimento tiroideu e orofaríngeo. A ecografia tiroideia não mostrou alterações. Ao exame objetivo da orofaringe, apesar de não terem sido detetadas alterações evidentes, em consulta de medicina dentária surge a suspeita de atresia do palato (palato ogival), confirmada com estudo complementar com ortopantomografia.
Ao longo de anos, o utente manteve acompanhamento regular em consulta com o MF, com uma periodicidade de cerca de dois meses no primeiro ano e, posteriormente, de seis em seis meses.
Este quadro clínico mostrou-se compatível com uma doença congénita caracterizada por malformações vertebrais, com possível associação a outras malformações viscerais. A tríade clínica é evidente neste doente. A maioria dos casos de SKF surge como mutação esporádica; todavia, estão descritos casos de hereditariedade autossómica dominante e recessiva.
Foi realizada avaliação familiar com genograma até três gerações, com o objetivo de identificar eventuais condições semelhantes; porém, não foram encontrados outros familiares com tais alterações. Crê-se, portanto, tratar-se de um caso de mutação esporádica. Não foi, contudo, até ao momento realizado estudo genético.
Atualmente, o utente tem dezoito anos, pretende ingressar no ensino superior, pratica exercício físico em contexto de aulas de educação física, sem queixas do foro osteo-articular.
Este processo de diagnóstico causou períodos de ansiedade na vida deste jovem e da sua família. Coube ao MF a gestão destes períodos através do seu acompanhamento periódico e regular. Foi sugerida eventual referenciação para acompanhamento por psicologia; contudo, o utente e a sua família preferiram manter apenas as consultas com o seu MF. Ao longo da marcha diagnóstica, o utente e a família foram sempre partes integrantes, mantendo-se informados de todas as suas suspeitas diagnósticas e das consequentes alterações identificadas.
Foram realizadas várias referenciações a consultas hospitalares durante este processo de diagnóstico, com o objetivo de descartar eventuais intervenções necessárias por parte dessas especialidades. O utente mantém acompanhamento nas especialidades de cardiologia e ortopedia, aguardando parecer desta última quanto a eventual necessidade de intervenção cirúrgica. Paralelamente, o MF assegura um seguimento regular do utente e do seu agregado familiar, monitorizando a evolução da síndrome e potenciais complicações decorrentes, de forma a garantir uma intervenção atempada e adequada. Contudo, prevê-se que, no futuro, as alterações estruturais osteo-articulares possam causar limitações funcionais ao nível da mobilidade deste utente, com consequente redução da sua qualidade de vida. Este processo tem sido discutido com o utente e com a sua família de forma a investir na reabilitação osteo-articular e na prevenção de eventuais comorbilidades futuras. Neste sentido, a manutenção de uma alimentação equilibrada com ingesta hídrica adequada, a prevenção do excesso de peso, a prática regular de exercício físico para reforço muscular e melhoria da mobilidade e o tratamento da dor são medidas fundamentais para reduzir a morbilidade associada à síndrome. Já a evicção de atividades que possam causar lesões é essencial para prevenir complicações, como fraturas, estenose vertebral, instabilidade cervical e disfunção articular.
O MF continua a prestar acompanhamento psicológico e social; porém, poderá ser novamente avaliada a necessidade de encaminhamento para apoio especializado no futuro. As questões estéticas e a adaptação social são temáticas abordadas pelo MF, não tendo sido motivo de preocupação até ao momento.
Comentário
Este caso clínico descreve o processo de diagnóstico de uma doença congénita complexa, com possível envolvimento multiorgânico. Trata-se de uma síndrome pouco reconhecida pela comunidade médica, o que pode condicionar atraso e subdiagnóstico desta condição. 1,4-6
Crê-se que a etiologia desta síndrome seja uma associação de fatores ambientais e genéticos que entre a terceira e a oitava semanas de gestação provoca um defeito no desenvolvimento embrionário, com resultado na formação anómala de vários órgãos e sistemas. 2,7
É de salientar o desempenho do MF no diagnóstico e suspeição desta condição clínica, tendo um papel chave na identificação das malformações osteo-articulares associadas a alterações cardíacas e renais. Estas alterações conjuntas levantaram a suspeição de uma síndrome congénita. Na presença de alterações osteo-articulares com fusão de vértebras cervicais, as hipóteses diagnósticas mais prováveis são malformações congénitas da coluna cervical ou patologias sistémicas, como a espondilite anquilosante, a artrite idiopática juvenil ou mesmo complicações de discites crónicas. Contudo, na presença de fusão de vértebras cervicais e dorsais associadas a malformações de arcos costais, craniofaciais, cardiovasculares e renais, a hipótese de SKF surge como a mais provável.
Importa notar, como dificuldade major, a articulação com a equipa de ortopedia, responsável pela orientação deste utente no que se refere à eventual intervenção cirúrgica. A incerteza quanto ao plano terapêutico causa sentimentos de angústia e preocupação a esta família. Neste sentido, o apoio do MF tem sido essencial para a gestão das emoções do utente e dos seus familiares.
O apoio contínuo e longitudinal fornecido pelo MF ao longo dos últimos quatro anos, focando-se não só na sua condição clínica, mas também no seu contexto psicossocial, tem sido fundamental à manutenção da homeostasia familiar. O MF tem exercido a função de moderador dos cuidados de saúde, realizando as referenciações necessárias e coordenando as várias especialidades envolvidas, tomando a responsabilidade central pela orientação e acompanhamento do utente e da sua família.
Agradecimentos
Um agradecimento especial à Dra. Magda A. Simões, pelo apoio e orientação na elaboração deste artigo.

