











 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
PermalinkIntrodução
A telangiectasia hemorrágica hereditária, também conhecida por síndrome Osler-Weber-Rendu é uma doença vascular hereditária autossómica dominante, que se apresenta com um conjunto de manifestações clínicas muito variadas, nomeadamente telangiectasias mucocutâneas, epistáxis, hemorragia gastrointestinal, anemia ferripriva e malformações arteriovenosas.1
Os estudos epidemiológicos apontam para uma prevalência estimada entre 1:5000 a 1:8000, com aproximadamente 85 000 casos a nível Europeu.2 Na literatura, estão descritas taxas de incidência superiores em determinadas áreas geográficas, nomeadamente, na população Afro-Caribenha, residente em Bonaire e Curaçau.3
Do ponto de vista fisiopatológico, trata-se de uma doença hereditária, tendo sido identificados dois genes, cuja mutação, condiciona esta patologia - gene da endoglina (ENG) e gene da activina (ALK-1).4
Para além da apresentação clínica mais frequente, esta patologia pode igualmente surgir com envolvimento pulmonar, hepático ou cerebral, através de malformações arteriovenosas.1
A telangiectasia hemorrágica hereditária é uma doença rara e com um espetro de manifestações clínicas muito variado, o que dificulta e atrasa o seu diagnóstico.1
O diagnóstico clínico precoce possível, a pesquisa de malformações arteriovenosas e o tratamento sintomático são aspetos fundamentais na abordagem destes doentes.1
O prognóstico destes doentes é variável e depende das manifestações da doença e a sua resposta à terapêutica instituída.1
Caso clínico
Os autores apresentam o caso clínico de um doente de 78 anos, género masculino, raça caucasoide, que recorreu ao Serviço de Urgência por clínica de dispneia para pequenos esforços, associado a astenia com algumas semanas de evolução.
Nos antecedentes pessoais, destacava-se um internamento prévio por insuficiência cardíaca descompensada, internamento na Unidade de Cuidados Intermédios de Gastroenterologia por hemorragia digestiva baixa (retorragias), tendo realizado estudo endoscópico sem alterações e videocápsula que evidenciou presença de sangue ao longo do intestino delgado. Referia igualmente episódios de epistáxis espontâneas desde a juventude. O doente negava história de alergias conhecidas.
Encontrava-se medicado com alopurinol 300 mg/dia, sinvastatina 20 mg/dia, pantoprazol 20 mg/dia, pentoxifilina 400 mg 2 vezes por dia, furosemida 40 mg/dia,
lisinopril 5 mg/dia.
Nos antecedentes familiares, desconheciam-se outros casos de hemorragias recorrentes ou outras patologias de relevo.
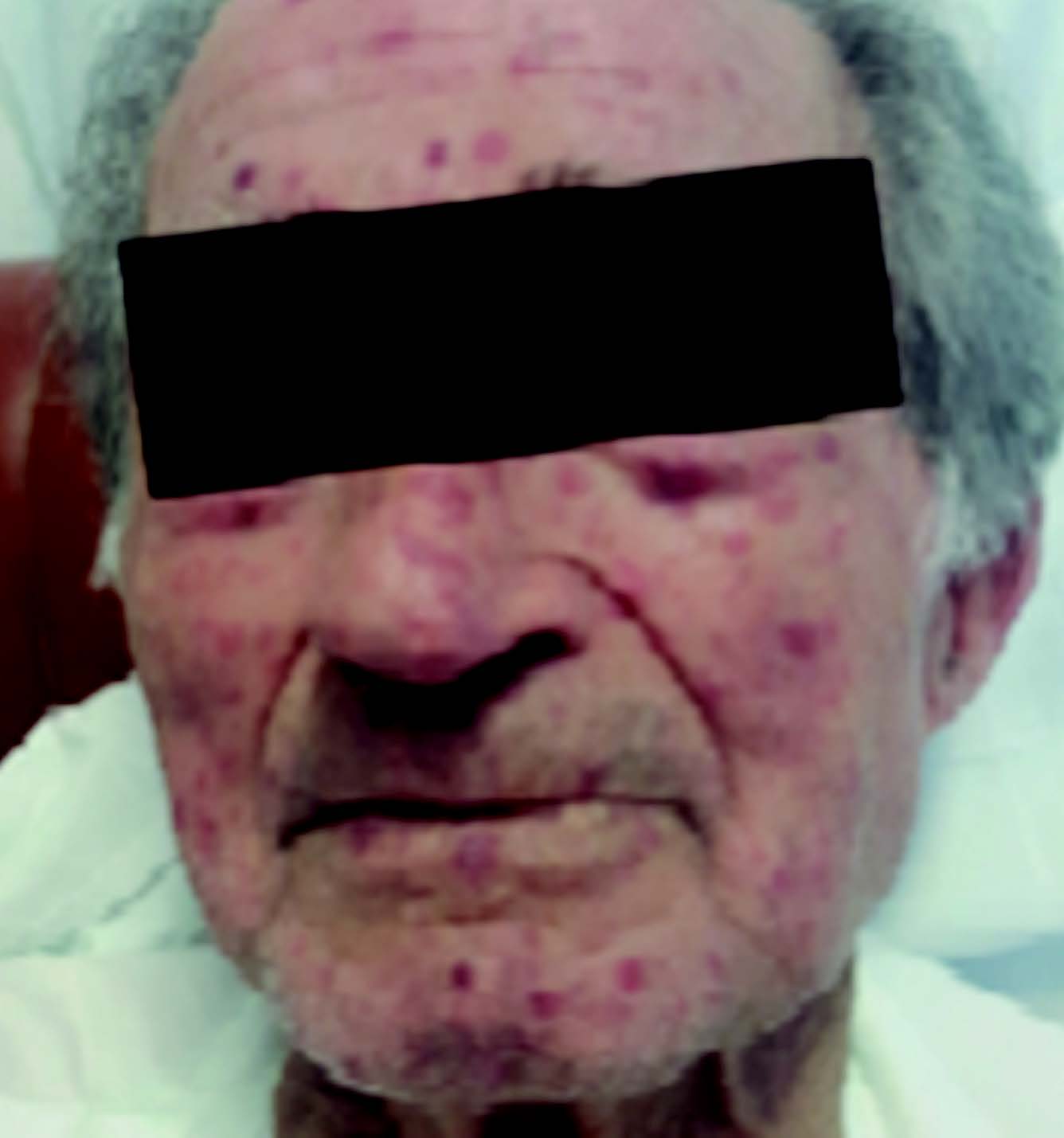
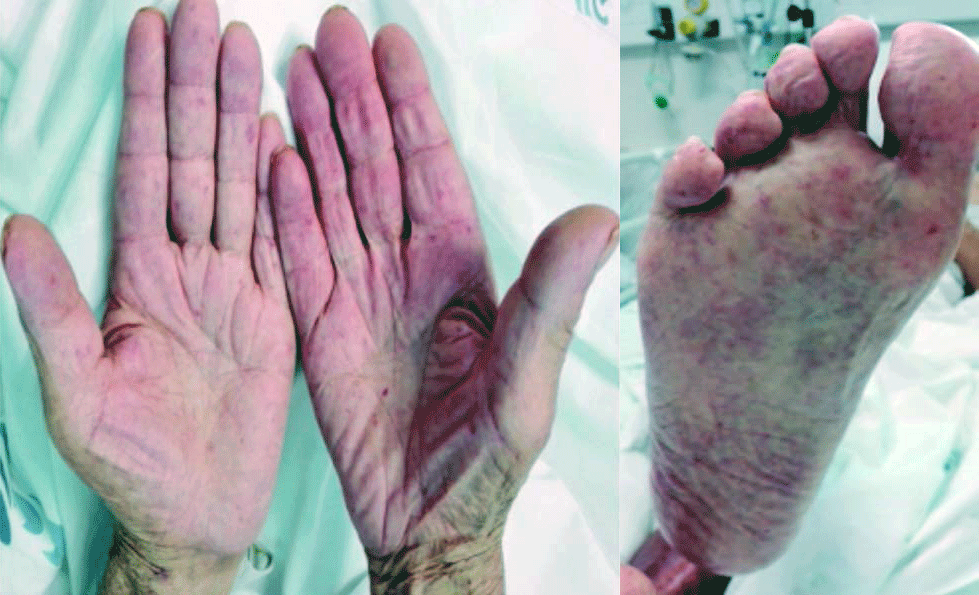
No exame objetivo, à admissão hospitalar, o doente apresentava-se consciente, orientado e colaborante, hemodinamicamente estável, eupneico em ar ambiente, com saturação periférica de O2 de 93%. A auscultação cardíaca, apresentava-se rítmico, com sopro sistólico grau III/VI audível no foco mitral; por outro lado, na auscultação pulmonar, evidenciava um murmúrio vesicular mantido bilateralmente, com crepitações bibasais. Do restante exame, destacava-se a presença de edemas bimaleolares e telangiectasias múltiplas na face, lábios, pirâmide nasal, membros superiores e inferiores.
Tabela 1: Estudo analítico à admissão hositalar.
| Resultados | Valores de Referência | |
|---|---|---|
| Hemograma | ||
| Leucócitos | 8 x 103/uL | 4 - 10,5 |
| Eritrócitos | 2,61 x106/uL | 4,7 - 6 |
| Hemoglobina | 7,7 g/dL | 13,5 - 18 |
| Hematócrito | 24,3% | 42 - 52 |
| VGM | 93 fL | 78 - 100 |
| HGM | 29,5 g | 27 - 31 |
| CHGM | 31,7 g/dL | 32 - 26 |
| Plaquetas | 211 x 103/uL | 150 - 450 |
| Bioquímica | ||
| TGO | 25 U/L | 0 - 38 |
| TGP | 15 U/L | 0 - 41 |
| FA | 166 U/L | 40 - 129 |
| GGT | 77 U/L | 8 - 61 |
| Bilirrubina total | 0,55 mg/dL | < 1 |
| Creatinina | 1,3 mg/dL | 0,7 - 1,2 |
| Estudo cinética ferro | ||
| Ferro | 24 ug/dL | 61 - 157 |
| Ferritina | 51,4 ng/mL | 21,81 - 274,66 |
| TIBC | 407 ug/dL | 149 - 491 |
VGM - volume globular médio; HGM - hemoglobina globular média; CHGM - concentração hemoglobina globular média; TGO - transaminase glutâmica oxalacética; TGP - transaminase glutâmica pirúvica; FA - fosfatase alcalina; GGT - gama glutamil transferase; TIBC - capacidade total de ligação ao ferro
Analiticamente, objetivou-se anemia normocítica com hemoglobina de 7,7 g/dL, com ferropenia (ferritina 51,4 ng/mL) (Tabela 1).
Realizou radiografia torácica póstero-anterior, verificando-se um aumento do índice cardiotorácico, reforço hilar bilateral e edema intersticial. O eletrocardiograma evidenciava um ritmo sinusal, com frequência cardíaca 70 batimentos/minuto, sem alterações do segmento ST ou alterações da repolarização.
Foi internado no Serviço de Medicina por insuficiência cardíaca descompensada e anemia com necessidade transfusional.
Nos primeiros dias de internamento, apresentou episódio de choque hipovolémico no contexto de epistaxe abundante não controlada e hematoquézias. Foi realizado suporte transfusional, dopaminérgico e ácido tranexâmico, no entanto, por persistência da clínica, foi transferido para observação pelas especialidades de Otorrinolaringologia (ORL) e Gastroenterologia do hospital central de referência.
Na observação por Otorrinolaringologia, foram visualizadas telangiectasias punctiformes. Para o controlo da epistaxes, o doente foi submetido a tamponamento nasal com merocel e gaze iodoformada anterior, bem como, eletrocauterização, com resolução clínica.
Por manter episódios de hematoquézias durante a permanência no Hospital Central, foi também observado pela Gastroenterologia. Foi submetido a endoscopia digestiva alta que não revelou alterações bem como colonoscopia com ileoscopia, que evidenciou presença de sangue no lúmen, no entanto, sem lesões sangrantes ou outras alterações visíveis. Em internamento prévio no Serviço de Gastroenterologia, o doente foi submetido a estudo do tubo digestivo através de videocápsula, o qual também não revelou alterações.
O doente permaneceu internado durante quatro dias na Unidade de Internamento de curta duração do Hospital Central para estabilização clínica, tendo sido transferido após 24 horas, para o nosso Hospital sem suporte aminérgico e sem novos episódios hemorrágicos, bem como estabilização da hemoglobina em 9,5 g/dL.
Durante o internamento no Serviço de Medicina, realizou tomografia computorizada abdominal, destacando-se hepatomegalia, sem lesões focais, fístulas artério-portais ou porto-venosas. Em ecocardiograma transtorácico, verificou-se uma boa função sistólica, regurgitação major da válvula tricúspide e hipertensão pulmonar com PSAP 58 mmHg.
Foi colocada a hipótese de síndrome Osler-Weber-Rendu, tendo sido solicitado estudo genético para pesquisa da mutação do gene da endoglina (ENG) e receptor quinase-like da activina (ALK-1), o que não foi aceite pelo doente.
O doente teve um internamento prolongado, complicado por intercorrências infeciosas e descompensações da insuficiência cardíaca.
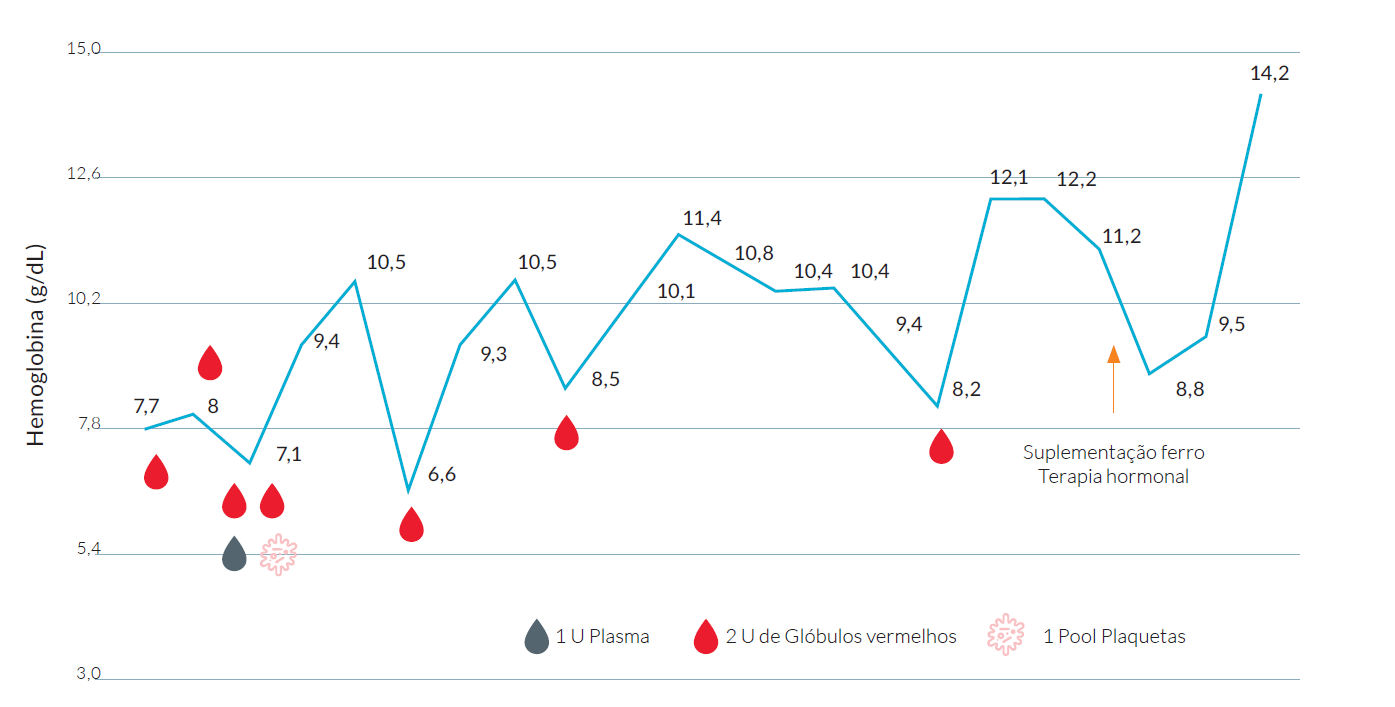
Em termos terapêuticos, para além do suporte transfusional e hemodinâmico, efetuou terapêutica com ferro e hormonoterapia com estradiol, tendo-se verificado estabilização dos valores da hemoglobina. (Fig. 1, evolução da hemoglobina)
Faleceu ao 30º dia de internamento, no contexto de edema agudo do pulmão.
Discussão e conclusão
Os critérios de diagnóstico de Curaçao baseiam-se em quatro achados clínicos: epistaxes espontâneas e recorrentes; telangiectasias mucocutâneas múltiplas; envolvimento visceral (malformações arteriovenosas, hemorragia gastrointestinal) e familiares em primeiro grau com esta patologia.5-6 O diagnóstico é definitivo se
o doente apresenta 3 ou mais destes critérios, suspeito no caso de apresentar 2 critérios e pouco provável se apenas apresentar um destes achados.5-6
O diagnóstico pode ser confirmado através da identificação das mutações genéticas ENG, ALK-1, no entanto, não são obrigatórias para se estabelecer o diagnóstico.5-6
O caso apresentado corresponde a uma telangiectasia hemorrágica hereditária, uma vez que, apresentava três critérios clínicos, epistaxes espontâneas e recorrentes, telangiectasias mucocutâneas e envolvimento visceral (hemorragia gastrointestinal). Durante o internamento, foram pesquisadas malformações arteriovenosas, nomeadamente a nível abdominal, não tendo sido objetivadas.
No ecocardiograma realizado para estudo de insuficiência cardíaca, foi diagnosticada hipertensão pulmonar. A hipertensão pulmonar encontra-se descrita nestes doentes como resultado do envolvimento pulmonar, nomeadamente por malformações.7
O tratamento desta patologia inclui terapias dirigidas, nomeadamente controlo de epistaxes (tamponamento nasal, terapia laser, dermoplastia septal para casos severos e refratários) e controlo de hemorragia gastrointestinal.1 Estão igualmente recomendadas terapêuticas sistémicas com o objetivo de reduzir os eventos hemorrágicos e que incluem o uso de ácido tranexâmico, terapias hormonais à base de estrogénios, tamoxifeno e bevacizumab.1 A Suplementação com ferro está recomendada em doentes com anemia ferripriva.1
A base teórica para o uso do ácido tranexâmico é a sua atividade antifibrinolítica, levando ao controlo da hemorragia. O ácido tranexâmico atua por inibição competitiva na ativação do plasminogénio, sendo análogo à lisina, e desta forma, impede a ligação da plasmina com a fibrina. Desta forma, a fibrina não será degradada, promovendo a formação de coágulo local. Este agente antifibrinolítico é geralmente bem tolerado, com poucos efeitos adversos descritos, sendo que, na maioria dos estudos verificou-se uma diminuição do número de episódios hemorrágicos, sobretudo de epistáxis.1
A terapêutica hormonal tem apresentado bons resultados no controlo de hemorragia gastrointestinal e mesmo de epistáxis nestes doentes, sendo que, o mecanismo subjacente parece ser a indução de metaplasia das mucosas, que resultam no espessamento do epitélio escamoso queratinizante, protegendo assim contra o trauma local.8
Outros fármacos estão a ser estudados, destacando-se o bevacizumab que é um anticorpo monoclonal humanizado IgG1 recombinante que se liga ao VEGF (vascular endothelial growth factor), que geralmente se encontra em elevada concentração nestes doentes.1 Ao ligar-se ao VEGF, neutraliza-o, sendo amplamente utilizado em esquemas de quimioterapia.1 Na literatura, este fármaco tem sido alvo de ensaios em doentes com esta síndrome, apresentando resultados promissores com redução dos episódios hemorrágicos. No entanto, não existe ainda uma indicação formal para o seu uso e os efeitos adversos descritos, nomeadamente hipertensão arterial, aumento do risco hemorrágico e trombótico, proteinúria, microangiopatia trombótica renal, perfuração gastrointestinal e do septo nasal, que não devem ser desprezados.1
O caso apresentado corresponde a um doente idoso, com degradação do seu estado geral ao longo dos vários internamentos, com maior grau de dependência e com diversas intercorrências, nomeadamente infeciosas, pelo que, em equipa, optou-se pela utilização de fármacos com mais estudos, maior evidência e com menos efeitos adversos descritos, nomeadamente, os agentes antifibrinolíticos e terapêutica hormonal, tendo o doente apresentado uma boa tolerância, sem efeitos adversos, mantendo ainda alguns episódios de epistaxes, no entanto, sem repercussões hemodinâmicas. Tal como, referido anteriormente, o doente veio a falecer ao 30º dia de internamento na sequência de complicações infeciosas e descompensação de insuficiência cardíaca.

