











 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introdução
A síndrome de Marfan é uma doença hereditária quase exclusivamente de transmissão autossómica dominante, estando descritos raros casos de transmissão recessiva. Apesar de na maioria dos doentes com síndrome de Marfan um dos pais ser afetado, esta pode resultar de uma mutação de novo em 25% ou mais dos casos.1-3 A incidência na população é de 1 em 3000-5000 indivíduos, não apresentando predomínio de género ou raça.2 É geralmente atribuída a uma mutação no gene codificante da fibrilina-1 - FBN1 (localizado no cromossoma 15), contudo, numa minoria dos casos (menos de 10%), esta mutação não é identificada. Na síndrome de Marfan, a síntese inadequada desta glicoproteína provoca alterações ao nível do tecido conjuntivo.1,2
O diagnóstico é baseado em achados clínicos e na história familiar, tendo como referência os critérios de Ghent revistos de 2010. Pela variabilidade fenotípica e, considerando que algumas manifestações têm penetrância dependente da idade, o diagnóstico poderá ser mais difícil em crianças e adolescentes.1,2,4,5
Com a adequada monitorização e tratamento, a sobrevida dos doentes com síndrome de Marfan é semelhante à da população em geral, sendo as doenças cardiovasculares as principais causas de morbilidade e mortalidade, principalmente a dissecção e a rotura da aorta.1,2,6,7
Descrevemos seguidamente a abordagem clínica e diagnóstico de um caso de síndrome de Marfan da nossa consulta do Centro de Saúde.
Caso clínico
Criança de 7 anos, sexo masculino, com antecedentes de perturbação de hiperatividade e défice de atenção, seguida em consulta de Pedopsiquiatria em regime privado e medicada com metilfenidato 27 mg.
Recorre ao centro de saúde, acompanhada pela mãe, para encaminhamento para a consulta externa de Pedopsiquiatria do hospital público da área de residência, tendo sido realizada consulta oportunista de vigilância de saúde infantil.
Na colheita da história clínica, não apresentava achados de relevo, com a exceção da referência, por parte da mãe, de que a criança era a mais alta da turma, com uma grande diferença relativamente aos seus colegas e da perceção de que esta apresentava descoordenação motora.
Sem outros antecedentes de relevo. Sem história de doenças hereditárias na família.
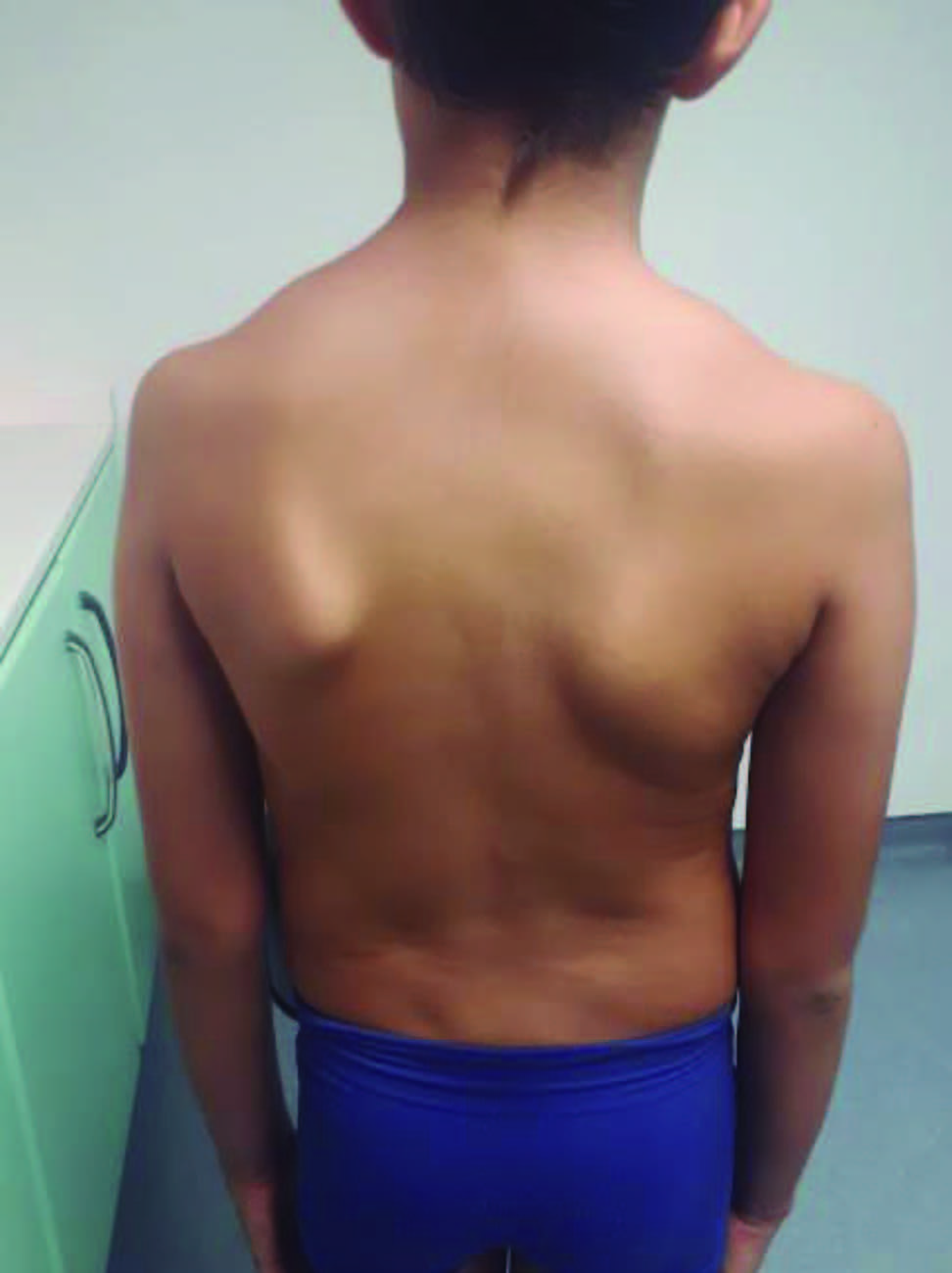
Ao exame objetivo, a criança apresentava 1,42 m de altura (superior ao percentil 97), pectus excavatum (Fig. 1), dedos das mãos longos e finos (aracnodactilia), pé plano (Fig. 2) e escoliose (Fig. 3).

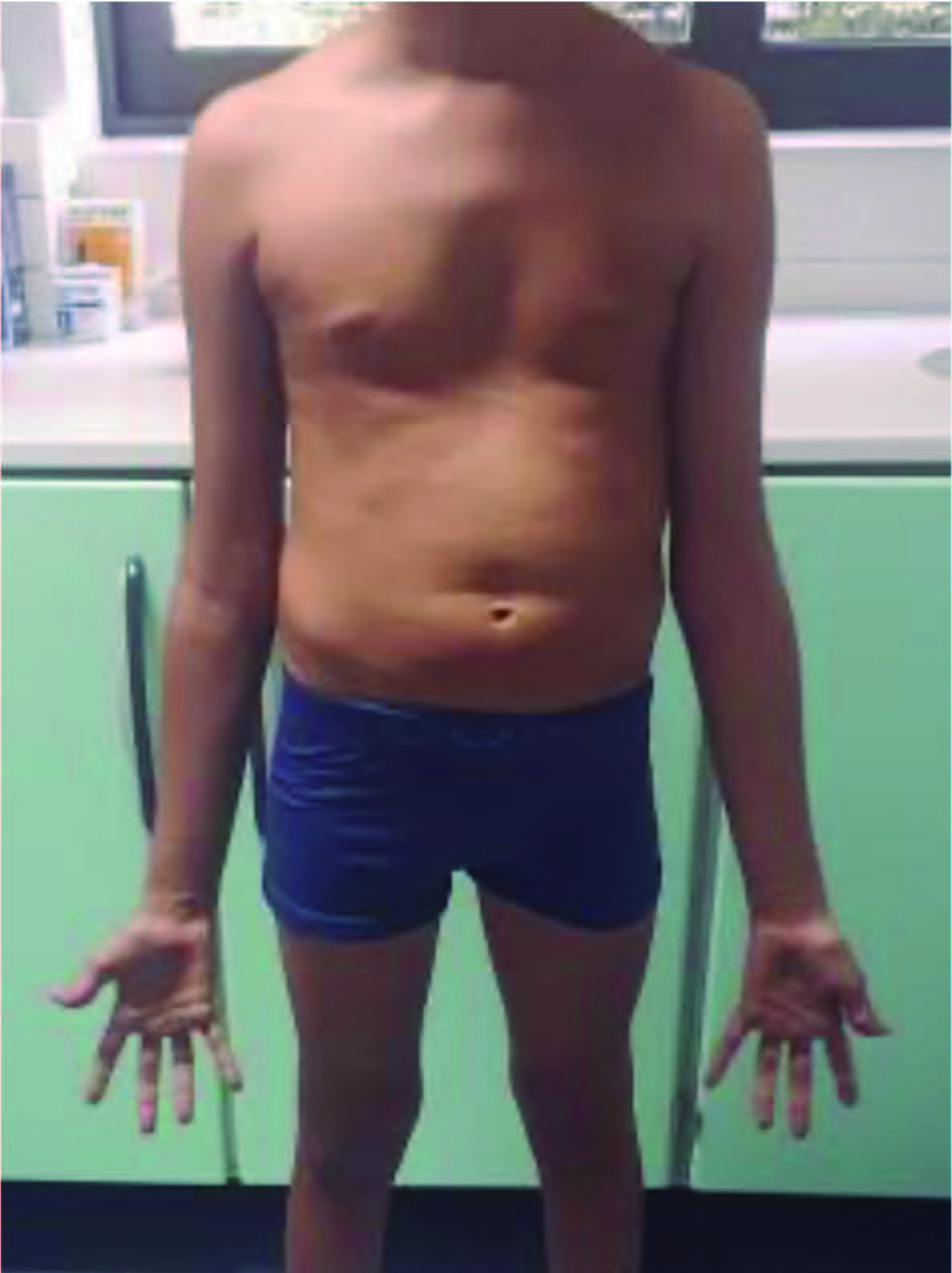
Figura 2: Visíveis as mãos do doente, observando-se a presença de dedos longos e finos (aracnodactilia) e presença de pé plano.
Apresentava um rácio do segmento superior/inferior reduzido (segmento inferior de 0,86 m, o que corresponde a um rácio de 0,65) e um rácio do comprimento dos membros superiores/altura aumentado (comprimento dos membros superiores de 1,54 m, com respetivo rácio de 1,08).
À auscultação cardíaca, de realçar a presença de um sopro sistólico de grau II/VI, audível no foco aórtico e sem irradiação.
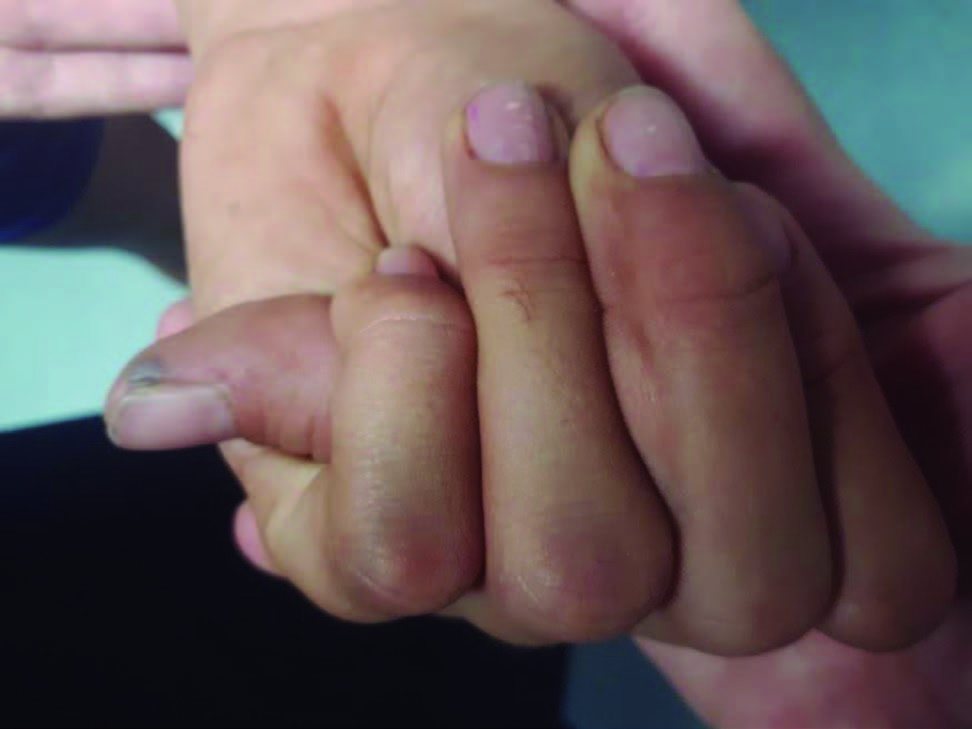
Figura 4: Sinal do polegar positivo - a falange distal do polegar ultrapassa o bordo cubital da região palmar após a adução do polegar contra a face palmar e flexão dos dedos sobre ele.
Por suspeita de doença do tecido conjuntivo, foram pesquisados o sinal do polegar (Fig. 4) e do punho, ambos positivos. O exame oftalmológico não revelou alterações.
Para o estudo das alterações detetadas no exame objetivo, foram solicitados exames complementares de diagnóstico, nomeadamente radiografia torácica, filme extralongo da coluna e ecocardiograma. O ecocardiograma mostrou a presença de um aneurisma da aorta ascendente (24 mm; 2,29 Z-score) e o filme extralongo da coluna corroborou a presença de escoliose. A radiografia torácica não evidenciou alterações.
Por suspeita de síndrome de Marfan, a criança foi referenciada para consulta hospitalar de Pediatria.
Na consulta hospitalar, confirmou-se a suspeita diagnóstica pelo cumprimento dos critérios de Ghent revistos, nomeadamente raiz da aorta igual ou superior a 2 Z-score e score sistémico igual ou superior a 7 (Tabelas 1 e 2), e a criança foi referenciada para as consultas de Oftalmologia, Ortopedia, Genética Médica, Cirurgia e Cardiologia Pediátricas.
Tabela 1: Critérios de diagnóstico definitivo de Ghent revistos da síndrome de Marfan (qualquer um dos seguintes critérios, tendo em conta a presença ou não de história familiar).
| Sem História Familiar | Com História Familiar Associada a |
|---|---|
| Raiz da aorta ≥+2 z-scoree ectopia do cristalino | Ectopia do cristalino |
| Raiz da aorta ≥+2 z-scoree mutação da Fibrilina-1 (FBN-1) | Scoresistémico ≥ 7 |
| Raiz da aorta ≥+2 z-scoreescoresistémico ≥ 7 | Raiz da aorta ≥ +3 z-score se < 20 anos de idade ou ≥ +2 z-score se > 20 anos |
| Ectopia do Cristalino e mutação da Fibrilina-1 |
Adaptado de Loeys BL, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476-85.8
Tabela 2: Cálculo do score sistémico para o diagnóstico de síndrome de Marfan.
| Característica | Pontos |
|---|---|
| Sinal do punho e do polegar | 3 |
| Pectus carinatum | 2 |
| Deformidade do retropé | 2 |
| Protusão acetabular | 2 |
| Ectasia da dura-máter | 2 |
| Pneumotórax | 2 |
| Sinal do punho ou do polegar | 1 |
| Pectus excavatum ou deformidade torácica | 1 |
| Pés planos | 1 |
| Escoliose ou cifose dorso-lombar | 1 |
| Rácio segmento superior/inferior diminuído e rácio comprimento dos membros superiores/altura aumentado* | 1 |
| Redução da extensão do cotovelo | 1 |
| Estrias cutâneas | 1 |
| Miopia | 1 |
| Prolapso da válvula mitral | 1 |
| Três das seguintes: dolicocefalia, fendas palpebrais oblíquas, enoftalmos, retrognatia e hipoplasia malar |
*Rácio segmento superior/inferior diminuído se < 0,95 (6-7 anos) e rácio comprimento dos membros inferiores/altura aumentado se ≥ 1,05
Discussão
A síndrome de Marfan é uma doença sistémica que pode manifestar-se fenotipicamente de várias formas. Apesar do avanço da genética, o seu diagnóstico continua a basear-se fundamentalmente nas manifestações clínicas e na presença de história familiar, assumindo, deste modo, a anamnese e o exame objetivo, um papel fulcral.1
A fibrilina-1 é a mais importante glicoproteína da matriz extracelular, contribuindo para a formação e manutenção das fibras elásticas.1,2 Neste caso clínico, a hiperlaxidez articular, que explica a possível descoordenação motora, associada à elevada estatura e à positividade do sinal do polegar e do punho, colocaram a síndrome de Marfan como o diagnóstico mais provável, apesar de raro.1,2,4,5 A ausência de história familiar, neste caso em particular, levanta a possibilidade de se tratar de uma mutação esporádica, o que torna o caso ainda mais raro.2
Após o diagnóstico de síndrome de Marfan, foi realizada consulta aos progenitores. Estes apresentavam uma estatura normal (inferior a 1,70 m) e não tinham qualquer alteração ao exame objetivo musculoesquelético e ocular. O ecocardiograma dos progenitores não revelou qualquer alteração. Foi ainda negada por ambos a existência de familiares com características fenotípicas da síndrome de Marfan.
O diagnóstico da síndrome de Marfan em idade pediátrica é, geralmente, dificultado pela ausência de algumas alterações sugestivas, que apenas se tornam evidentes na idade adulta.2 Contudo, o diagnóstico precoce é crucial, uma vez que possibilita uma orientação e vigilância adequadas destes doentes, melhoria do seu prognóstico e redução do risco de complicações, nomeadamente osteoarticulares, oftalmológicas e cardiovasculares.2 Relativamente a estas últimas, a orientação por parte da Cardiologia Pediátrica é fundamental, tendo em conta que o risco de dissecção da aorta é um dos principais motivos de morbilidade e mortalidade, podendo requerer terapêutica farmacológica (como beta-bloqueantes) ou, em casos mais graves, intervenção cirúrgica, nomeadamente a cirurgia eletiva da raiz da aorta.2,7
Sendo esta uma doença de transmissão autossómica dominante, com penetrância elevada,2 o estudo genético, apesar de não ser obrigatório para o diagnóstico, é importante para orientar a terapêutica e a frequência da vigilância, na identificação de familiares potencialmente afetados, assim como, futuramente, no diagnóstico pré-natal.7
A evicção de desportos de contacto, do exercício isométrico e do mergulho em profundidade é essencial para a proteção dos doentes com síndrome de Marfan.3 Devido à hiperlaxidez articular, a realização de exercícios que promovam o reforço muscular e consequente estabilização articular é, de igual forma, importante, permitindo aumentar a funcionalidade dos doentes e evitar as complicações a longo prazo.3
No que concerne ao prognóstico destes doentes, se adequadamente vigiados e tratados, a sobrevida aproxima-se da população em geral.6,7
Este caso clínico pretende alertar para a importância do reconhecimento desta síndrome que se encontra subdiagnosticada, principalmente em idade pediátrica. A exploração das alterações encontradas ao exame objetivo possibilitou o diagnóstico precoce da síndrome de Marfan e, consequentemente, uma maior celeridade na orientação e instituição da terapêutica, com provável impacto no prognóstico da doença.

