











 Portuguese (pdf)
Portuguese (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Similars in
SciELO
Similars in
SciELO 
 Permalink
Permalink
Introdução
A granulomatose com poliangeíte (GcP) anteriormente designada granulomatose Wegner (GW) é uma vasculite necrotizante granulomatosa sistémica, mediada por fatores imunes. O diagnóstico é baseado nos critérios clínicos, radiológicos, sorológicos e anatomopatológicos propostos pelo American College of Rheumatology.1
A base do tratamento é a corticoterapia combinado ao uso de imunossupressores. A GcP é rara e potencialmente grave, com uma taxa de mortalidade e morbilidade importantes, quer pela própria doença, quer pelos efeitos secundários da terapêutica.
Caso Clínico
Doente do sexo masculino, caucasiano, 42 anos, com antecedentes de diabetes mellitus não insulino-tratado e tabagismo. Internado em dezembro 2017 no serviço de Psiquiatria, por episódio maníaco inaugural com sintomas psicóticos. Exames de diagnóstico preliminares (hemograma, bioquímica com perfil hepático, renal e ionograma, e eletrocardiograma) sem alterações de relevo, tomografia computorizada (TC) cranioencefálica à admissão com “sinusopatia etmoido-maxilar de predomínio esquerdo”. Durante o internamento foi solicitada colaboração da Medicina Interna por queixas constitucionais.
O doente referia tosse não produtiva com 6 meses de evolução, rinorreia e hiperemia conjuntival sazonal. Referia poliartralgia de ritmo inflamatório há mais de 15 anos, associada a rigidez matinal. Do exame objetivo, apenas ao nível da semiologia pulmonar se verificou aumento da transmissão das vibrações vocais direitas com submacicez à percussão e fervores dispersos.
Analiticamente, anemia normocítica normocrómica (hemoglobina 10 g/dL). Proteína C reativa de 14 mg/L (N: 0,0-8,0 mg/L); velocidade segmentar (VS) 67 mm.
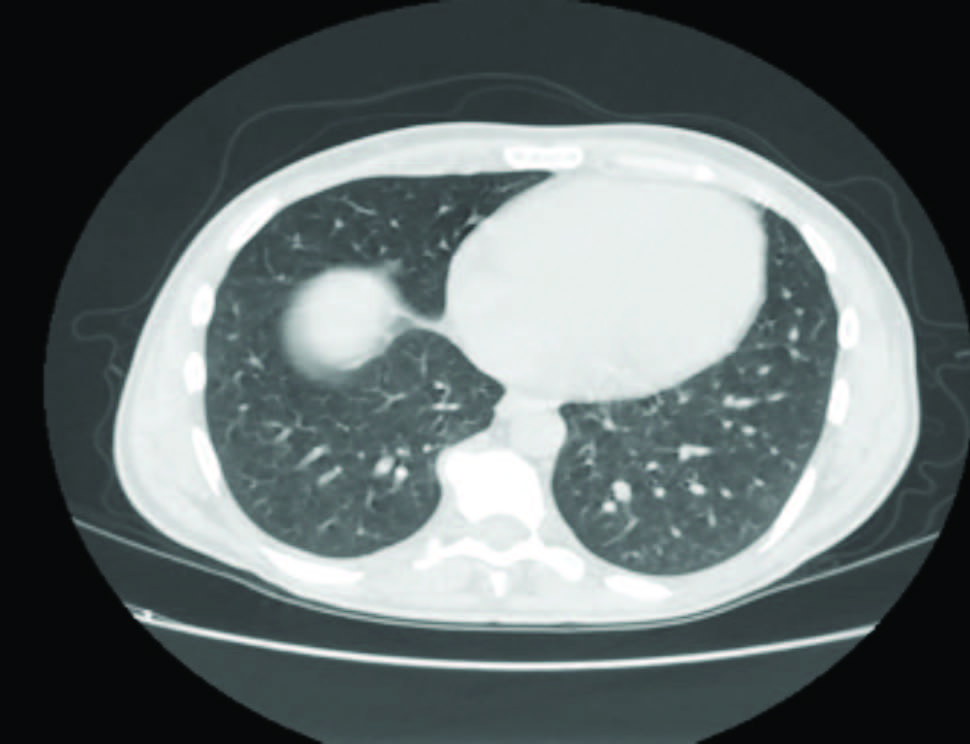
Figura 1: Tomografia computorizada do tórax, com hipotransparência justa-hilar direita de cerca de 3x1,8 cm e vários micronódulos dispersos no parênquima pulmonar.
Radiografia de tórax com hipotransparência justa-hiliar direita. TC torácica: “Observa-se ectasia com espessamento parietal brônquico difuso e diversos micronódulos distribuídos por ambos os campos pulmonares, sendo o maior de cerca de 1 cm” (Fig. 1). Face ao resultado da TC de tórax, foi solicitado avaliação da Pneumologia, e realizou biópsia percutânea guiada por TC, complicado com pneumotórax e posteriormente com empiema pulmonar por Streptococcus anginosus com insuficiência respiratória global e necessidade de suporte ventilatório não invasivo temporário.
Realizou estudo imunológico, destacando-se, anticorpos anticitoplasmáticos, determinados por imunofluorescência indireta e método de ELISA, anti-proteinase 3 (cANCA) positivos (25 U/mL, normal < 5 U/mL).
A anatomia patológica do nódulo pulmonar veio a revelar: “fragmentos de parênquima pulmonar com fibrose irregular… denso infiltrado inflamatório misto com esboço de granulomas; vasos de pequeno e médio calibre com lesões de vasculite, necrose fibrinoide e orla histiocitária. Aspetos compatíveis com GcP”. Ainda na marcha diagnóstica, optou-se por realizar ressonância magnética cranioencefálica (RM-CE) que revelou: “… Lesão cortical frontal superior esquerda com foco típico de disrupção da barreira hemato-encefálica/status vasculítico agudo, em contexto de mais provável granuloma não caseoso - poliangeíte granulomatosa necrotizante”. De acordo com critérios clínicos e imunológicos, foi estabelecido diagnóstico de GcP com envolvimento do trato respiratório e cerebral. Foi estabelecido plano terapêutico com ciclofosfamida 500 mg 15/15 dias, durante 6 ciclos e prednisolona 1 mg/kg/dia. Durante a terapêutica, manteve trimetoprim/sulfametoxazol (960 mg/dia). Após instituição da terapêutica, o doente evolui favoravelmente sem evidência de complicações iatrogénicas, mantendo vigilância clínica regular em consulta de Medicina Interna, Psiquiatria e Doenças Autoimunes.
Discussão
A granulomatose com poliangeíte (GcP) é uma vasculite sistémica granulomatosa que atinge vasos de pequeno e médio calibre. A GcP pertence ao grupo das vasculites necrosantes (juntamente com poliangeíte microscópica, poliarterite nodosa clássica e síndrome de Churg-Strauss). É uma doença rara, com uma prevalência estimada de cerca de 3 /100 000 indivíduos, sem prevalência de género. Afeta predominantemente caucasianos, entre os 40-55 anos.2 Embora a etiopatogénese da GcP não esteja ainda estabelecida, é certo que mecanismos de autoimunidade estão implicados.2-5 Os principais órgãos afetados são: vias aéreas superiores, pulmões e rins.
A apresentação clínica é inespecífica, normalmente o doente apresenta sintomas constitucionais, como astenia, perda ponderal, mialgias, artralgias. Pode existir febre, que pode ser decorrente da atividade da própria doença ou resultado de uma infeção secundária. Achados laboratoriais na GcP incluem: elevação da VS e fator reumatoide, anemia, trombocitose, leucocitose.
O envolvimento das vias aéreas superiores ocorre em 95% dos pacientes, particularmente nos seios paranasais, e os sinais desse acometimento são: rinorreia e ulceração da mucosa nasal, podendo ocorrer perfuração do septo nasal. A biópsia revela inflamação e formação de granulomas, com ou sem vasculite. O envolvimento pulmonar está presente em cerca de 90% dos pacientes, com tosse, dispneia e hemoptises. Tipicamente, aparecem infiltrados cavitários múltiplos, bilaterais e nodulares,2 e é importante fazer-se o diagnóstico diferencial com histoplasmose pulmonar, comum em imunocomprometidos. A doença renal, presente em até 80% dos pacientes, é responsável pela maioria das mortes decorrentes da GcP. O acometimento renal caracteriza-se por uma glomerulonefrite focal e segmentar, que pode evoluir para uma glomerulonefrite rapidamente progressiva. A microscopia eletrónica e por imunofluorescência não revelam evidências de deposição de imunocomplexos nas lesões renais da GcP, ao contrário das outras formas de glomerulonefrite.
Outros órgãos podem ser acometidos, como olhos (52% dos pacientes, podendo apresentar conjuntivite, episclerite); pele (46% dos pacientes, podendo manifestar púrpura palpável, úlceras ou nódulos subcutâneos); coração (8% apresentam pericardite ou miocardiopatia). O envolvimento do sistema nervosa central (SNC) na GcP é incomum, sendo as vasculites cerebrais a forma mais frequente de acometimento, mas faltam estudos amplos sobre envolvimento do SNC.6-8 As manifestações neurológicas incluem, paralisia, convulsões e sintomas neuropsiquiátricos. Os achados de vasculite cerebral são inespecíficos e melhor visualizados por RM-CE. A biópsia geralmente não é possível, tornando o diagnóstico de vasculite cerebral, na GcP um desafio.
Recentemente, percebeu-se a forte associação entre a existência de níveis elevados de ANCA e a GcP.2,3,5,9 Aproximadamente, 70%-90% dos pacientes com GcP ativa apresentam o cANCA positivo e níveis elevados parecem ter uma elevada sensibilidade e especificidade quer no diagnóstico, quer como marcador da atividade da doença, permitindo antever as recidivas da doença.2,3,5,10 A utilização do método de ELISA para quantificação ANCA parece ser superior à imunofluorescência indireta para o imunodiagnóstico e follow-up da doença.9 Outras vasculites, nomeadamente a poliarterite microscópica e a síndrome de Churg-Strauss, cursam com valores de ANCA elevados, daí a importância da clínica e histologia para o diagnóstico diferencial.2,3 Pela heterogeneidade da apresentação clínica, a GcP coloca problemas de diagnóstico diferencial.2,5
Pacientes com manifestações clínicas compatíveis deveriam ser submetidos à biópsia tecidual, sendo que o tecido pulmonar oferece alta positividade diagnóstica. Contudo, o diagnóstico histológico nem sempre é obtido, uma vez que as alterações anatomopatológicas típicas têm uma distribuição irregular nos tecidos e por vezes não é pertinente equacionar procedimentos invasivos nestes doentes, dada a gravidade da doença e a necessidade de iniciar rapidamente terapêutica.5
A classificação das vasculites sistémicas permaneceu controversa nos últimos anos. Dois sistemas de classificação principais foram propostos por um longo tempo - as definições da Conferência de Consenso de Chapel Hill (CHCC) e os critérios da American College of Rheumatology de 19901 (Tabela 1).
Tabela 1: Critérios de GcP, recomendações da Academia Americana de Reumatologia.
| 1 | Inflamação nasal ou oral (úlceras orais dolorosas, ou não, ou corrimento nasal sanguinolento |
| 2 | Radiografia de tórax anormal (nódulos, infiltrados fixos ou cavidades) |
| 3 | Sedimento urinário anormal (micro-hematúria ou cilindros he máticos) |
| 4 | Inflamação granulomatosa em biópsia (a histologia deve mos trar inflamação granulomatosa em parede arterial, região perivascular ou extravascular de atérias e arteríolas) |
Os doentes que apresentam pelo menos dois destes critérios podem ser diagnosticados como portadores de GcP, com sensibilidade e especificidade, respetivamente, de 88,2% e 92%. Estes critérios apresentavam várias limitações, como não incluir ANCA como um critério potencial. Mais recentemente surgiu o algoritmo da Agência Europeia de Medicamentos (EMEA), destina-se à classificação em vez do diagnóstico de casos e inclui, serologia ANCA positiva e evidência de vasculite na biópsia. Uma lista de recursos substitutos para uso em apoio ao diagnóstico de GcP é fornecida, incluindo marcadores radiológicos, histológicos e clínicos.11
A avaliação da atividade e do dano é um componente essencial, as escaladas mais usadas são o Birmingham Vasculitis Activity Score (BVAS / BVAS V3.0) e o Vasculitis Damage Index (VDI).12
O tratamento da GcP passa pelo uso de prednisolona inicialmente na dose de 1 mg/kg/dia durante 1 mês, e posteriormente com descalada lentamente durante cerca de um ano2 e ciclofosfamida na dose de 2 mg/kg/dia. A ciclofosfamida deve ser continuada por 1 ano após a indução da remissão completa. Em casos de resistência ao tratamento (sem melhoria passadas 4 semanas ou 6 semanas com melhoria < 50% segundo BVAS) utiliza-se o rituximab. A plasmaferese pode ser ainda equacionada nos casos refratários e em GcP rapidamente progressivas. O metotrexato e a azitioprina (2 mg/kg/dia) podem ser utilizados como uma alternativa à ciclofosfamida quando existirem contraindicações ou elevada toxicidade a este fármaco.2,5 Cerca de 75% dos doentes atinge a remissão completa com a terapêutica.2,5 Após a remissão
da doença, é fundamental manter um follow-up apertado. Efetivamente, cerca de 50% dos doentes apresenta uma recaída num período de 5 anos 2,5,10 e 42% apresenta complicações tardias relacionadas com a terapêutica, em particular com a ciclofosfamida,2 verificando-se uma incidência aumentada de carcinoma da bexiga, doenças linfoproliferativas e neoplasias cutâneas, num período que pode atingir os 15 anos após a terapêutica imunossupressora.2 A utilização de trimetoprim/sulfametoxazol na GcP, na dose de 800/160 mg/dia durante 24 meses, parece reduzir a incidência de recaídas pelo seu papel na prevenção de infeções e uma ação anti-inflamatória pela interferência no stress oxidativo na patogénese da GcP.2,10 A GW é uma patologia potencialmente grave,2,5,10,12 com uma taxa de mortalidade de cerca de 13% e com uma morbilidade importante.2,5,12 Como já foi referido, a avaliação seriada dos ANCA durante o follow-up dos doentes com GcP é um bom método preditivo das recaídas da doença.2,10
No caso clínico apresentado, o doente apresentou-se com alterações neuropsiquiátricas e sintomas constitucionais. Face aos achados imagiológicos e a positividade dos cANCA realizou estudo histológico que foi compatível com o diagnóstico. O doente tem evoluído favoravelmente, sem fatores de morbilidade associados à doença ou à toxicidade dos fármacos utilizados, e esperamos a remissão completa da doença.

