










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
versão impressa ISSN 2341-4545
GE Port J Gastroenterol vol.27 no.4 Lisboa ago. 2020
https://doi.org/10.1159/000504760
CLINICAL CASE STUDY
Whipple’s Disease: A Rare Cause of Malabsorption Syndrome
Doença de Whipple: uma causa rara de síndrome de má absorção
Joana Cardosoa, Lídia Gomesb, Sandra Santosa, Hélder Moreirac, Paula Gomesd, João Ruaa, Jorge Fortunaa
aDepartment of Internal Medicine, Coimbra University and Hospital Centre, Coimbra, Portugal; bDepartment of Pneumology, Coimbra University and Hospital Centre, Coimbra, Portugal; cDepartment of Anatomic Pathology, Coimbra University and Hospital Centre, Coimbra, Portugal; dDepartment of Imagiology, Coimbra University and Hospital Centre, Coimbra, Portugal
* Corresponding author.
ABSTRACT
Introduction: Whipple’s disease is a rare, chronic, systemic disease caused by the actinomycete Tropheryma whipplei. Clinical manifestations vary widely depending on the affected system, the most common being the digestive tract. Case Presentation: The authors report the case of a 52-year-old man with malabsorption syndrome, diarrhea, marked weight loss, melanoderma, and visual and proprioception disorders. Periodic acid-Schiff staining of a proximal small bowel biopsy and peripheral-blood PCR identification of T. whipplei confirmed the disease. The patient was initially treated with intravenous ceftriaxone, followed by oral trimethoprim/sulfamethoxazole with significant clinical improvement. Conclusions: This case is reported due to its rarity and the diagnostic challenge it presents. Although uncommon, Whipple’s disease should be considered as a differential diagnosis of malabsorption syndrome due to its systemic impact and possible treatment with targeted antibiotic therapy.
Keywords: Whipple’s disease, Tropheryma whipplei, Periodic acid-Schiff
RESUMO
Introdução: A doença de Whipple é uma doença rara, crónica, sistémica, causada pelo actinomicete Tropheryma whipplei. As manifestações clínicas variam de acordo com o órgão envolvido, sendo a sua apresentação típica predominantemente associada ao sistema digestivo. Caso clínico: Os autores descrevem um caso de um homem de 52 anos com síndrome de má absorção, diarreia, perda de peso marcada, melanodermia, bem como alterações visuais e a nível da propriocepção. A coloração com PAS na biopsia de intestino delgado proximal e a identificação por PCR no sangue periférico confirmou a doença. Iniciou antibioterapia com ceftriaxone EV, seguida de trimetropim/sulfametoxazol oral com recuperação clínica significativa. Conclusão: Este caso é descrito pela sua raridade e pelo desafio diagnóstico. Apesar de incomum deve ser considerada como diagnóstico diferencial do síndrome de má absorção atendendo ao seu impacto sistémico e ao possível tratamento com antibioterapia dirigida.
Palavras-Chave: Doença de Whipple, Tropheryma whipplei, Ácido periódico-Schiff
Introduction
Whipple’s disease (WD) is a chronic systemic disease caused by the Gram-positive bacillus Tropheryma whipplei. It is a rare disease (0.5–1 cases per million) first described in 1907 by George Hoyt Whipple as an intestinal lipodystrophy [1].
Recent studies have shown that T. whipplei is a commensal bacterium and not an obligate pathogen [2, 3]. Genetic predisposition and immunological host factors have been implicated in the pathogenesis of this infection.
Men are more frequently affected than women, and the age of onset of the first symptoms is around 40 years of age. The classic presentation of the disease begins with arthralgia followed by diarrhea, weight loss, fever, lymphadenopathies, and occasionally neurological, cardiac, or ocular manifestations [3].
The diagnosis is a challenge because it is a disease with systemic repercussions and nonspecific symptoms. The disease is confirmed by duodenal biopsies characterized by foamy macrophages in the lamina propria, whose cytoplasm contains large amounts of particles that are periodic acid-Schiff (PAS) positive.
The most common treatment regimen includes a 2-week course of intravenous cephalosporin followed by oral sulfamides for 1–2 years. Generally, long-term antibiotic treatment can relieve symptoms and cure the disease; if left untreated, it is progressive and fatal. Prognosis may improve with earlier recognition, diagnosis, and treatment [4].
Case Report/Case Presentation
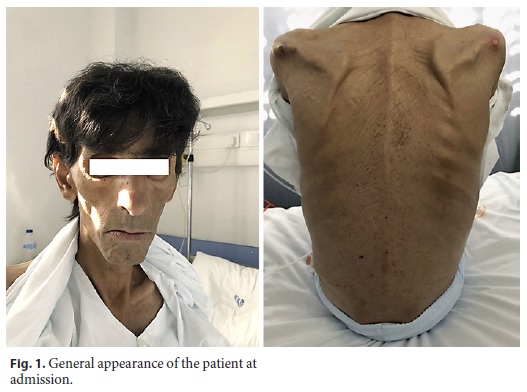
A 52-year-old man, with multinodular goiter and heavy smoking (40 pack-years) and drinking habits (50 g/day), reported a history of abdominal pain, myalgias, anorexia, and asthenia for 2 years, worsening in the last 6 months with watery diarrhea, without mucus or blood but with poorly digested food, as well as marked abdominal distension and 30% body weight loss (40 kg at admission). He denied association of the diarrhea with the consumption of any specific food, namely dairy or gluten-containing food, or previous episodes of prolonged diarrhea. Once inquired he further mentioned bilateral sacroiliac arthralgia and xerostomia especially in the prior 6 months. He denied nausea, vomiting, fever, night sweats or chills, respiratory or genito-urinary symptoms. The patient was currently unemployed but had worked for several years in agriculture and pork industry. His social background was precarious with inadequate hygiene conditions. He denied recent travel or contact with infected patients and was only medicated with iron supplements. Physical examination showed hypotension (blood pressure: 90/65 mm Hg), normal heart rate (78/min), apyrexia (axillary temperature: 36.0 ° C), cachexia, infracentimetric cervical and supraclavicular adenopathies, generalized diminished muscular strength, pale mucosae, and melanoderma (Fig. 1). Cardiac auscultation revealed normal heart sounds, without murmurs or bruits. Pulmonary auscultation was unremarkable as well. Neurological examination revealed an impaired visual acuity, mainly nocturnal, disturbed proprioception, an aquilian reflex abolished, reduction of patellar reflexes, and dysmetria in the finger-to-nose test.
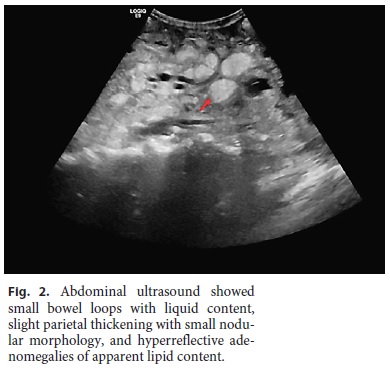
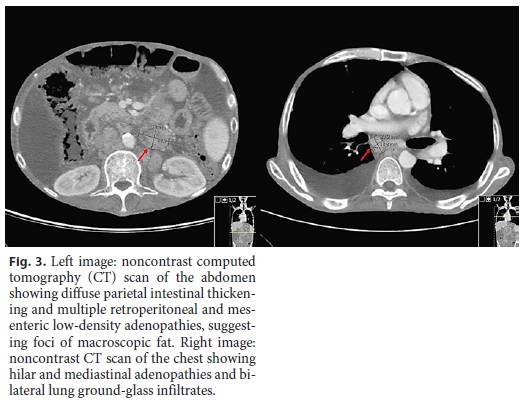
Laboratory tests showed normocytic anemia (Hb 8.5 g/dL, normal [N]: 13.0–17.5), lymphopenia (0.39 × 103/μL, N: 1.0–3.0), profound hypokalemia (2.06 mmol/L, N: 3.5–5.1), hypocalcemia (1.62 mmol/L, N: 2.10–2.55), and hypophosphatemia (0.42 mmol/L, N: 0.81–1.45), hypoproteinemia with severe hypoalbuminemia (16 g/L, N: 35–50), very low cholesterol levels (total cholesterol 27 mg/ dL), and marked deficiency of vitamins A (0.036 mg/L, N: 0.260– 0.890) and D (4.3 ng/mL, N: > 29). Erythrocyte sedimentation rate and C-reactive protein were slightly increased (20 mm/h and 4.6 mg/dL, respectively). Fecal analysis showed positive calprotectin (129 mg/kg, N: < 50 mg/kg) and steatocrit > 50%, confirming an inflammatory diarrhea with steatorrhea, consistent with malabsorption. Stool elastase was low (70 mg/g, N: > 200), probably due to high water stool content, but this value did not allow the exclusion of pancreatic insufficiency. An abdominal ultrasound showed small bowel loops with liquid content, slight parietal thickening with small nodular morphology, and hyperreflective adenomegalies of apparent lipid content, very suggestive of WD (Fig. 2). A chest-abdomen-pelvis computed tomography scan (Fig. 3) revealed hilar and mediastinal adenopathies and bilateral lung ground-glass infiltrates and confirmed diffuse parietal intestinal thickening and multiple retroperitoneal and mesenteric low-density adenopathies, suggesting foci of macroscopic fat. No signs of digestive tract neoplasm were observed. Extensive workup was undertaken to exclude other possible causes, such as parasitic infections (3-day fecal ova and parasite test), tuberculosis (interferon-γ test, blood and fecal culture, and polymerase chain reaction [PCR]), human immunodeficiency virus and cytomegalovirus infection, celiac disease and inflammatory bowel disease, hyperthyroidism, and lymphoproliferative diseases (peripheral blood smear and immunophenotyping).
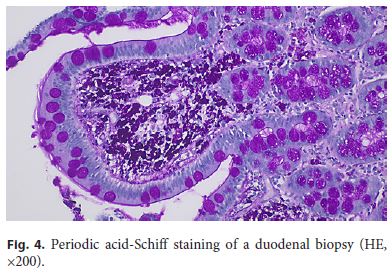
Finally, a digestive endoscopy was performed revealing thickened duodenal folds, multiple lymphangiectasia, and pale mucosa, whose biopsies revealed dilated villi with expanded lamina propria due to numerous foamy macrophages with clear and granular eosinophilic cytoplasm (PAS-D-positive granules) (Fig. 4). A transthoracic echocardiogram excluded relevant abnormalities, and a cerebrospinal fluid PCR for T. whipplei was negative. Peripheral blood detection of T. whipplei DNA by PCR further supported the diagnosis of WD.
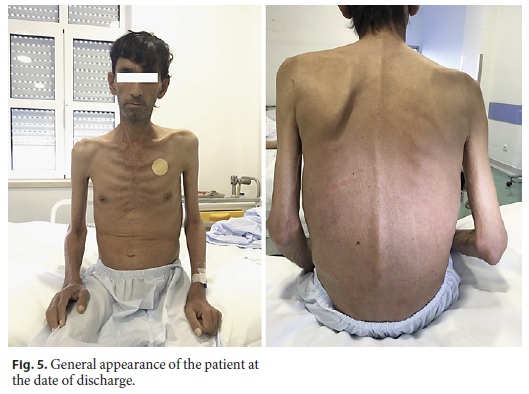
The patient was treated with intravenous ceftriaxone (2 g/day) for 2 weeks and supportive therapy, including parenteral nutrition and correction of ionic and vitamin deficiencies. During this period, the patient showed progressive clinical improvement, with increasing tolerance to enteric nutrition, regulation of intestinal transit, and weight regain (8 kg in 2 weeks) (Fig. 5). At discharge, treatment was shifted to oral trimethoprim/sulfamethoxazole (800/160 mg b.i.d.) with the intention of a 1-year treatment course. He was discharged to a rehabilitation facility, presenting at the 1-month follow-up appointment fully asymptomatic with a total weight regain of 16 kg since admission and full mobility capacity restored (Fig. 6).
Discussion/Conclusion
In 1907, Whipple first described pathological findings in the small intestine and mesenteric ganglia of patients with malabsorption syndrome and named it intestinal lipodystrophy as he suspected it to be a disease of lipid metabolism [1]. This hypothesis was later abandoned after the discovery that the observed macrophages contained glycoproteins instead of lipids, as evidenced by their PAS positivity [4, 5]. Only in 1961, after electron microscopy identification of bacilliform bodies in the cytoplasm of the macrophages, was it possible to confirm the bacterial etiology of this disease [6].
Due to its rarity, little is known about WD pathophysiology. However, some studies have identified T. whipplei in saliva samples, gastric juice, and duodenal biopsies from healthy individuals, suggesting its ubiquitousness in the digestive tract [4, 5]. Pathogenicity appears to be related to immunological host factors and genetic susceptibility, suggested by association of the disease with HLAB-27 [7].
WD is undoubtedly a rare disease. A multicenter study [8] evaluated the number of cases described over 15 years and reported the presence of only 33 cases. More recent studies indicate an annual global incidence of 12 cases [9]. WD predominantly affects Caucasian men with a mean age of 50 years at the time of diagnosis [2–4, 8]. The prevalence is higher among populations with poor personal hygiene, such as the homeless, and among professionals in contact with soils and feces, such as farmers and sewer workers [9].
Clinical manifestations are diverse and sometimes nonspecific. Although classic disease remains the most common presentation, clinical descriptions have identified different forms of presentation. The following forms have been described: acute infection, chronic disseminated disease (or classic WD), and chronic localized infection.
T. whipplei has been detected in several acute infections. The percentage of symptomatic people at the time of primary infection is unknown. Cases of pneumonia, acute gastroenteritis, and bacteremia have been described [9, 10].
The typical clinical manifestation of WD is a malabsorption syndrome with weight loss, diarrhea, abdominal pain, fever, and lymphadenopathy [3, 8–11]. Diarrhea is watery and sometimes steatorrheic, leading to severe weight loss and cachexia [10]. Joint involvement is very frequent, usually intermittent, migratory, oligo- and polyarticular. It often precedes the diagnosis about 10 years [3]. The most serious manifestation of WD is central nervous system involvement, since irreversible damage can persist despite adequate and successful treatment. About 10–40% of patients present with neurological manifestations, such as dementia, supranuclear ophthalmoplegia, altered level of consciousness, or the almost pathognomonic signal of oculomasticatory myorhythmia and oculofacioskeletal myorhythmia [10]. Cardiac manifestations (endocarditis, myocarditis, and pericarditis) are also common [10].
Chronic localized infection has been described after evidence of the disease without histological intestinal identification. In this form of WD, there is no systemic involvement and the potential for relapse is not the same as in classic WD [9]. The most common presentation is bacterial endocarditis with persistently negative blood cultures, mostly diagnosed by histology or PCR of the extracted valve. Lymphatic (mesenteric lymphadenopathy), articular (isolated synovitis), and ocular (uveitis) involvement has also been described, as well as primary central nervous system infection [9, 10].
Recently, a new subtype associated with immunosuppression has been identified [10]. It is believed that the use of immunosuppressive therapy alters the physiological balance of the intestinal barrier in chronic carriers, leading to pathogenesis. Anti-TNF agents are the most frequently associated drug class, raising concern for a possible increase in the number of cases with their widespread use.
The main differential diagnoses of the typical form are other malabsorptive diseases with small intestinal involvement (celiac disease, tuberculosis, and Mycobacterium avium infection of the small bowel, histoplasmosis, and Crohn’s disease) and infiltrative diseases of the small intestine (non-Hodgkin lymphoma and amyloidosis) [11, 12]. Other causes of diarrhea, such as hyperthyroidism and pancreatic insufficiency, should also be excluded.
Anemia (usually microcytic or normocytic), relative lymphopenia, and increased erythrocyte sedimentation rate are consistent findings. Laboratory markers of malnutrition are usually found, such as decreased total lymphocyte count, and low serum albumin and transferrin. Steatorrhea is often severe in advanced WD [11].
Abdominal ultrasound, while not specific, may reveal suggestive findings, such as dilated and thickened small bowel wall, with the disappearance of normal wall stratification and brilliant hyperechoic luminal contents mixed with a few hypoechogenic parts, and diffusely echogenic retroperitoneal lymphadenopathy, sometimes with concomitant free abdominal fluid or lymph node cavitation [13].
Upper endoscopy with intestinal biopsy and PCR analysis are the most effective methods of investigation. The diagnosis is based on positive PAS staining of duodenal biopsies; however, this can be positive in other circumstances, such as Mycobacterium, Rhodococcus equi, or Bacillus cereus infection [9–11, 13, 14]. Immunohistochemical analysis using specific antibodies allows the direct visualization of bacteria in samples with greater sensitivity and specificity than those of PAS staining [15].
WD was an invariably fatal disease until 1961, when evidence of its infectious etiology allowed treatment through antibiotic therapy [14]. Currently, trimethoprim/ sulfamethoxazole is considered the mainstay of treatment for a duration of 12 months. If the disease is more severe and/or affects the central nervous system, intravenous treatment with ceftriaxone 2 g per day for 15 days is recommended, followed by oral treatment with trimethoprim/ sulfamethoxazole. Occasional resistance to sulfamethoxazole has been described and has been associated with treatment failure and clinical relapse [16]. Doxycycline and hydroxychloroquine association has been suggested as an alternative. Relapses can occur as late as 20 years after the initial diagnosis and may occur in other organs than those previously involved [9]. Some series have reported that 9.1–15% of patients with classic WD developed a failure or a relapse during or after treatment with trimethoprim/sulfamethoxazole [17].
After a transient clinical improvement with antibiotic therapy, patients can present with fever and arthralgia probably related to an immune reconstitution syndrome. This phenomenon is thought to occur in about 10% of patients. After exclusion of an infectious cause, corticosteroids should be started early [3].
Early and proper recognition of the disease can lead to full cure with long-term antibiotic therapy. Extraintestinal symptoms often disappear within a few days, and gastrointestinal symptoms and malnutrition are resolved within 2–3 months [11]. Neurologic manifestations may be irreversible despite treatment.
The follow-up of patients must be extended to careful life-long monitoring for any relapse of the disease. In patients with gastrointestinal symptoms, duodenal samples should be assessed histologically and with PCR; however, PAS staining in biopsies may remain positive for years [2].
Although rare, WD should be suspected in patients with classic and nontypical symptoms of WD since it can be fatal if undiagnosed and untreated. The reported patient showed compatible symptoms; the hyperechogenic adenomegalies observed in the ultrasound further suggested the diagnosis, and the additional tests led to confirmation of WD and its successful treatment.
References
1 Morgan AD. The first recorded case of Whipple’s disease? Gut. 1961 Dec;2(4):370–2.
2 Schneider T, Moos V, Loddenkemper C, Marth T, Fenollar F, Raoult D. Whipple’s disease: new aspects of pathogenesis and treatment. Lancet Infect Dis. 2008 Mar;8(3):179–90.
3 Puéchal X. Enfermedad de Whipple. EMC - Tratado de Medicina [Internet]. 2019 Feb;23(1):1–6.
4 Sellin J, Beales IL. Whipple’s Disease: A Well-Done Outcome to a Rare Disease. Dig Dis Sci. 2019 Jan;64(1):9–11.
5 La Scola B, Fenollar F, Fournier PE, Altwegg M, Mallet MN, Raoult D. Description of Tropheryma whipplei gen. nov., sp. nov., the Whipple’s disease bacillus. Int J Syst Evol Microbiol. 2001 Jul;51(Pt 4):1471–9.
6 Ehrbar HU, Bauerfeind P, Dutly F, Koelz HR, Altwegg M. PCR-positive tests for Tropheryma whippelii in patients without Whipple’s disease. Lancet. 1999 Jun;353(9171):2214.
7 Dobbins WO 3rd. HLA antigens in Whipple’s disease. Arthritis Rheum. 1987 Jan;30(1):102–5.
8 Hujoel IA, Johnson DH, Lebwohl B, Leffler D, Kupfer S, Wu TT, et al. Tropheryma whipplei Infection (Whipple Disease) in the USA. Dig Dis Sci. 2019 Jan;64(1):213–23.
9 Fenollar F, Lagier JC, Raoult D. Tropheryma whipplei and Whipple’s disease. J Infect. 2014 Aug;69(2):103–12.
10 Crespo Ramírez E, Lemus Saracino A, Gonzáles Pérez S. Enfermedad de Whipple, una causa poco frecuente de diarrea. Rev Cienc Méd. 2017 Jun;21(3):133–7.
11 Bure J, Kopáčová M, Douda T, Bártová J, Tom J, Rejchrt S, et al. Whipple’s Disease: Our Own Experience and Review of the Literature. Gastroenterol Res Pract. 2013;2013:478349.
12 Pimentel-Nunes P, Cardoso E, Brandão C, Duarte HR, Lobo C, Henrique R, et al. Apresentação da Doença de Whipple como Linfoma. J Port Gastrenterol. 2011 May;18(3):135–8.
13 Fraquelli M. Lymphangiectasia, Whipple’s Disease, Eosinophilic Enteritis. In: Maconi G, Bianchi Porro G, editors. Ultrasound of the Gastrointestinal Tract. Berlin, Heidelberg:Springer;2012. pp. 129–33.
14 Carneiro C, Lima P, Barbosa I, Chaves F. Doença de Whipple: um desafio diagnóstico. Acta Med Port. 2004 Jan;17:481–6.
15 Prudent E, Le Guenno G, Jonckheere S, Vankeerberghen A, Lepidi H, Angelakis E, et al. Fluorescent in situ hybridization can be used as a complementary assay for the diagnosis of Tropheryma whipplei infection. Infection. 2019 Apr;47(2):317–21.
16 Fenollar F, Perreal C, Raoult D. Tropheryma whipplei natural resistance to trimethoprim and sulphonamides in vitro [Internet]. Int J Antimicrob Agents. 2014 Apr;43 (4):388–90.
17 Lagier JC, Fenollar F, Lepidi H, Raoult D. Failure and relapse after treatment with trimethoprim/sulfamethoxazole in classic Whipple’s disease. J Antimicrob Chemother. 2010 Sep;65(9):2005–12.
Statement of Ethics
For this study, the patient’s written informed consent to a description of his case and publication of photographs was obtained, although the patient’s anonymity was maintained. This study did not require approval from the appropriate ethics committee.
Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Sources
The authors have no funding sources to declare.
* Corresponding author.
Joana Cardoso
Department of Internal Medicine, Hospital Geral
Centro Hospitalar e Universitário de Coimbra, Quinta dos Vales
São Martinho do Bispo 108, PT–3041-801 Coimbra (Portugal)
E-Mail jbernardinocardoso@gmail.com
Received: July 22, 2019; Accepted after revision: October 27, 2019

