











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkHistorical Background
Celiac disease (CD) is a systemic disease triggered by gluten ingestion, in genetically predisposed individuals. It manifests as an autoimmune enteropathy associated with specific circulating autoantibodies and human leukocyte antigen haplotype (HLA-DQ2 or HLA-DQ8) [1].
Aretaeus of Cappadocia, in 250 A.D., described a chronic perturbation of “pepsis” (i.e., digestion) and “anadosis” (i.e., absorption) resulting in a general debility which was named “celiac diathesis” [2, 3]. The word “celiac” is derived from the Greek “koiliakos,” which means abdominal [4]. However, it was only in 1888 that Samuel Gee [5] published the first modern clinical detailed description of CD. In 1908, in the USA, Christian Herter [6] published a similar description, emphasizing the retardation in growth. For several years, CD was known as Gee-Herter disease [7].
Diet was proposed as a causal contributor since Arateus. In the twentieth century, several diets were advocated, such as the banana diet [8] and the Fanconi diet based on fruits and vegetables [7]. The causal link to grain consumption (wheat, rye, barley, and, to a lesser extent, oats) was described in the forties by the Dutch pediatrician Willem-Karel Dicke. This link came from the observation of the effect of food scarcity on children with CD during the Second World War. Dicke observed that symptoms of children with CD improved when they were not eating bread or grains and worsened after the war ended and these foods reentered their diet [7, 9].
Paulley [10], in 1954, described detailed histological anomalies in the small bowel from surgical specimens (chronic inflammation and atrophy) from patients with CD.
In 1964, Berger et al. [11] reported the presence of serum anti-gliadin antibodies (AGA) in CD. It took up to 20 years for serology be considered a diagnostic criterion [12, 13]. More sensitive and specific serological tests have been identified since then [14, 15].
In 1972, Falchuk et al. [16] described the association between a specific HLA genotype and CD and hypothesized that CD is a consequence of carrying an abnormal immune response gene to gluten. We know now that HLA-DQ2/8 is necessary for the development of CD, making HLA determination the third pillar in the diagnosis of CD [17].
Epidemiology
The prevalence of CD varies according to age, gender, and region. A recent meta-analysis estimated a global prevalence of 1.4% by serological tests and 0.7% by intestinal biopsy [18]. This is probably an underestimation of the real prevalence of CD, since it is estimated that only 1 in 5 patients with CD is diagnosed [19]. In some regions, such as Asia and Africa, the number of reported cases of CD is extremely low, even though wheat consumption is increasing [20] and the frequency of CD-associated HLA alleles seems similar to that in Western countries [21]. As such, in Asia, CD is likely to be even less efficiently diagnosed, with less awareness for the asymptomatic forms of the disease [22]. In this millennium, the prevalence of CD seems to have increased by 33%, for unknown reasons, but it is probably associated with environmental factors [18].
Women are 1.5 times more afflicted than men [18]. The incidence of CD is approximately 2 times more frequent in children than in adults, with a second peak in incidence between 50 and 69 years [23].
Genetics are a main factor in the risk for CD, with over 40 genetic loci associations besides the HLA-DQ2/8 haplotype [24].
Environmental factors have been studied as risk factors for CD [25-27]. The association between age at gluten introduction and CD is controversial [28-32]; however, current recommendations advise gluten introduction between 4 and 12 months of age [33]. The amount and pattern of gluten consumption may have a role and may account for the different prevalence rates of CD across Europe [34].
Breastfeeding does not seem to protect from CD [28, 32, 35-37]. Recurrent respiratory infections (in infants) and gastrointestinal infections (rotavirus and adenovirus in children and Campylobacter in adults) seem to be associated with CD [25, 26, 38], though the evidence is weak [39].
Lastly, high-risk groups for CD include first-degree relatives of CD patients [40] (with a prevalence up to 7.5%) [41], and patients with type 1 diabetes mellitus (T1DM) [42] or other autoimmune diseases [43], IgA deficiency [44], and chromosomopathies such as Down syndrome [45] and Turner syndrome [46].
Pathophysiology
CD results from an intense immune response to gluten leading to small bowel injury with consequent malabsorption and autoimmune phenomena [47].
Gluten consists of a group of proteins from Gramineae of the Triticiae tribe, particularly wheat, rye, and barley. Oats are phylogenetically more distant (Aveneae tribe) but share sufficient similarities to induce symptoms in some patients. Rice, maize, sorghum, and millet are distant enough not to trigger CD [48]. Gluten is the Latin word for “glue,” owing its name to its viscoelastic and adhesive properties [26]. The word gluten is widely used to refer to disease-inducing Gramineae proteins; however, strictly speaking, gluten specifically refers to proteins from wheat. Similar proteins in rye are secalins, and in barley they are hordein [49]. Wheat gluten contains 2 major protein components, i.e., monomeric water-soluble gliadins and multimeric water-insoluble glutenins [50].
Gluten peptides are highly enriched in proline and glutamine. Gastrointestinal proteases are deficient in prolyl-endopeptidase activity. As such, the high-proline content makes gluten resistant to gastrointestinal cleavage, allowing the subsistence of polypeptides with up to 33 amino acids [51]. This increases gluten’s immunogenicity, since major histocompatibility complex (MHC) II molecules only present peptides at least 9 amino acids long [52].
Gluten peptides are not freely absorbed and need a disruption in the epithelial barrier to translocate into the lamina propria, where antigen-presenting cells (APC) reside. That may occur through a damaged epithelium, induced by transient intestinal infection, drug-induced inflammation (e.g., nonsteroidal anti-inflammatory drugs), or dysbiota-induced disassembly of enterocyte tight junctions. Alternatively, gluten can cross the epithelium in a transcellular pathway through binding of gluten-secretory IgA complexes to the transferrin receptor CD71 or inside dendritic cells that cross the epithelium [17, 50, 53-55].
MHC-II molecules bind preferentially to peptides with negatively charged amino acids. Even though gluten peptides have very few charged amino acids, they are highly susceptible to deamidation on their glutamine residues to negatively charged glutamate by tissue transglutaminase (tTG) [52]. Deamidation significantly increases the stability of the gluten-MHC complex, increasing their immunogenicity [56].
APC present deamidated gluten peptides bound to a specific MHC class II that map to the HLA-DQ locus. That locus codifies antigen-presenting glycoproteins that are heterodimers constituted by a α-chain (encoded by the DQA1 allele) and a β-chain (encoded by the DQB1 allele). Only HLA-DQ2.5, DQ2.2, and DQ8 (and probably DQ7.5) bind to deamidated gluten peptides, and so their presence is necessary for the development of CD. However, it is not a sufficient condition, since these haplotypes are also found in 40% of the general population [53, 57]. Nonetheless, about 90% of CD patients express HLA-DQ2.5 and roughly 10% express HLA-DQ2.2 or HLA-DQ8 [58]. HLA-DQB1*02, present in HLA-DQ2.5 and HLA-DQ2.2, confers a higher risk for CD compared to the presence of HLA-DQB1*03, present in HLA-DQ8. The frequency of HLA-DQB1*02 seems to be even higher in CD patients with T1DM [59]. Furthermore, there is a dose effect for HLA-DQ2.5, since homozygous individuals have a 5-fold increased risk for CD and for severe disease [60]. Haplotypes, HLA, and their risk are shown in Table 1.
APC (dendritic cells and macrophages) present the complex MHC-II-deamidated gliadin to CD4+ T cells, inducing a proinflammatory phenotype in T cells [61]. Activated CD4+ T cells promote differentiation of B cells into plasma cells and release proinflammatory cytokines such as interferon-γ and interleukin-21 that activate intraepithelial CD8+ T cells [17, 58]. Interleukin-15 further promotes intraepithelial CD8+ T cell differentiation into a cytotoxic NK cell-like phenotype, damaging enterocytes [61]. Activated CD4+ T cells also secrete tumor necrosis factor-α, which acts on intestinal fibroblasts inducing their secretion of: (1) matrix metalloproteinases (contributing to mucosal destruction by dissolution of connective tissue) [62] and (2) epithelial mitogen keratinocyte growth factor (contributing to crypt epithelial cells hyperplasia) [63].
Lastly, tTG-deamidated gluten complex may bind to receptors allowing internalization into specific B cells, which then act as APC to CD4+ T cells, further fueling the immune response. Conversely, those B cells may differentiate into plasma cells, explaining why specific anti-tTG antibody production only occurs under a gluten-containing diet [64]. The pathophysiology of CD is summarized in Figure 1.
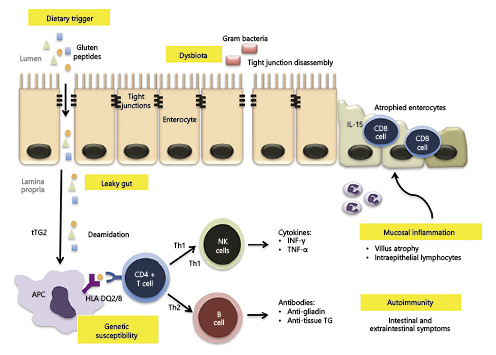
Fig. 1 Pathogenesis of CD. Gluten peptides reach the lamina propria through a leaky gut, with increased intestinal permeability, which may be due to drug-induced inflammation or dysbiota-induced disassembly of enterocyte tight junctions. Gluten peptides are then deamidated by tTG, which allows them to be presented to CD4-T cells by APC that exhibit MHC class II molecules encoded by the haplotypes HLA-DQ2 or HLA-DQ8. This will trigger T-helper 1 and T-helper 2 immune responses that result in mucosal cytotoxicity and inflammation, as well as autoimmune extraintestinal phenomena.
Clinical Manifestations and Associated Diseases
The clinical spectrum of CD is broad, which accounts for the challenging diagnosis. Classical gastrointestinal manifestations are more common in children, whereas adults tend to be paucisymptomatic [25]. Classical manifestations are chronic diarrhea (in 35%), abdominal pain (28%), and weight loss (22%) [53, 65]. CD can also present paradoxically with chronic constipation (20%), abdominal distension (20%), gastroesophageal reflux (12%), and even obesity [25, 53].
The most common extraintestinal manifestations are decreased bone mineralization (osteopenia in 50-70% and osteoporosis in 5.5% of cases), anemia (32%), arthralgia (29%), fatigue (26%), and neurological symptoms (20%), particularly gluten ataxia and peripheral neuropathy [53, 66]. Gluten ataxia is an autoimmune injury of the cerebellum, induced by gluten ingestion, which manifests with a typical serology and abnormal gait, muscle coordination, and fine control of voluntary movements, as well as cerebellum atrophy on magnetic resonance imaging (up to 60%). The mean age at onset is around 50 years [67] and the effect of a gluten-free diet (GFD) is controversial [68-71]. Gluten neuropathy is a sensitive neuropathy that is associated with serological evidence of CD, which initially affects the hands and feet but usually progresses. The mean age at diagnosis is 55 years and a GFD can improve symptoms regardless of the presence or absence of enteropathy [71].
CD can also manifest with hypertransaminasemia (9-14%), recurrent aphthous stomatitis, tooth enamel defects, infertility, delayed puberty, and a short stature [53, 66, 72]. Most extraintestinal manifestations improve with a GFD, but an early diagnosis is crucial and some manifestations, such as enamel defects, may be irreversible [66].
CD is associated with many genetic disorders. The prevalence of CD is higher in patients with chromosomopathies (i.e., 5-10% in patients with Down, Turner, and Williams syndromes) [73]. This might be explained by the proinflammatory millieu and impaired function of CD4+ T cells associated with chromosomopathies [46, 74].
Autoimmune glandular diseases, particularly T1DM and thyroid disease, are strongly associated with CD; 10-30% of CD patients have 1 of those 2 autoimmune diseases and up to 7% of patients with autoimmune glandular diseases have CD [25, 75]. In fact, those conditions share a genetic background with a tight link to HLA-DQ2/8 and DR3/4 [75]. Interestingly, the prevalence of autoimmune diseases increases with increasing age at diagnosis, probably as a consequence of a higher duration of exposure to gluten [76].
Herpetiform dermatitis (HD) is a dermatological autoimmune disease that also shares a genetic background with CD. Up to 20% of CD patients develop HD and more than 90% of HD patients have CD. HD diagnosis can be confirmed by skin biopsy demonstrating IgA deposits in the papillary dermis adjacent to the lesion. These patients present anti-tTG as well as IgA anti-epidermal transglutaminase antibodies. HD responds to GFD, although transient treatment with dapsone may be needed [25, 66, 77].
CD patients have an increased risk of hepatic diseases such as steatosis, autoimmune hepatitis, primary biliary cholangitis (at least a 20-fold increase) [78], and primary sclerosing cholangitis (4- to 8-fold increase) [79].
Finally, patients with a selective IgA deficiency present a risk of CD that is 10-20 times higher [80]. The reverse is also true, i.e., IgA deficiency is 10-15 times more frequent in patients with CD [81].
Diagnosis
Who to Test?
Current guidelines recommend testing for CD patients with signs, symptoms, or laboratorial evidence of malabsorption, unexplained fatigue, and recurrent mouth ulcers. Furthermore, patients with T1DM or autoimmune thyroid disease should be regularly tested [82, 83]. Importantly, the presence of symptoms is not required for a diagnosis of CD [84].
CD screening is recommended in patients with irritable bowel syndrome, since these patients are 4 times more likely to have CD than the general population, even patients presenting with obstipation [85]. Lastly, first-degree relatives of CD patients should be screened, though there are no recommendations regarding the time interval for rescreening [73].
The following high-risk groups should also be considered for screening: children and adolescents with chromosomopathies and patients with metabolic bone disorders, unexplained neurological symptoms, hypertransaminasemia or infertility, and dental enamel defects [73]. CD screening recommendations are displayed in Table 2.

Population-based screening is not recommended, since it has not been proven that the diagnosis of asymptomatic patients improves their quality of life [87, 88].
Diagnostic Tools
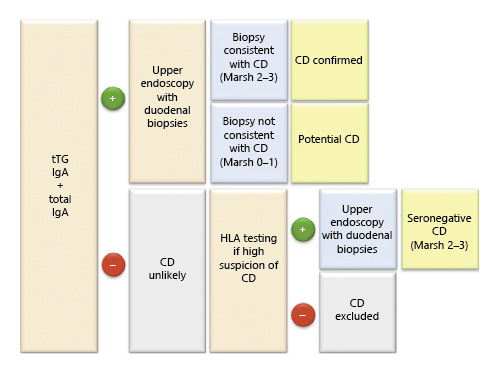
CD diagnosis relies on 3 main pillars, i.e., serological tests, duodenal histology, and genetic testing [89]. The diagnostic algorithm is presented in Figure 2.
Anti-tTG IgA is the recommended first-line serological test, as it is the most sensitive test (98%) and has a very good specificity (96%) [47]. Anti-tTG is determined through ELISA, allowing quantitation. Anti-endomysial (EMA) IgA reacts to the same antigen of tTG, but bound to tissue, requiring immunofluorescence in tissue from primate oesophagus or human umbilical cord. As such, EMA is more expensive, technically more challenging, and operator dependent, and it only allows qualitative results [90]. The anti-EMA IgA test is the most specific serological test [91] and it should be used as a confirmatory test, especially when the anti-tTG level is lower than 2 times the upper limit of normal [83, 89]. Anti-tTG and anti-EMA IgG have a low sensitivity and should be interpreted carefully [91].
AGA is not recommended for CD diagnosis due to the low sensitivity and specificity [82, 92]. More recently, anti-deamidated gliadin peptides (DGP), which are assessed by an ELISA assay, have been adopted, particularly IgG that is superior to other IgG antibodies (88% sensitivity and 99% specificity) [91]. Anti-DGP IgG is particularly useful in patients with a selective IgA deficiency [83, 93].
Diagnosis should start with measurement of both anti-tTG IgA and IgA serum levels. Positive anti-tTG IgA should prompt duodenal biopsies to confirm the diagnosis. When the anti-tTG IgA titer is low (4-10 U/mL) with normal IgA levels, anti-EMA IgA should be assessed. Lastly, when IgA <1 mg/dL, IgG antibodies should be assessed (particularly anti-DGP IgG) [90].
All serological tests should be performed under a gluten-containing diet to avoid false-negative results [82]. False-positive results may occur with intestinal infections (e.g., Giardia lamblia) [94], chronic liver disease [95], congestive heart failure [96], and hypergammaglobulinemia [97]. Of note, serological tests have demonstrated high intertest and interlaboratory variability at lower ranges, and this should be confirmed by alternative tests when levels are lower than 10 times the upper limit of normal [84].
All patients with positive serological tests should undergo an upper endoscopy with duodenal biopsies [73]. Endoscopy is also indicated in patients with negative serological tests when the clinical suspicion is high [82].
Endoscopic findings alone present a sensitivity that ranges from 11 to 22% for CD [98]. Some findings, however, are very specific (up to 99%) for mucosal atrophy, i.e., scalloping duodenal folds, fissuring, and a mosaic pattern of the mucosa [99, 100]. Less specific findings are duodenal erosions, loss of folds, nodular mucosa, and enhanced submucosal vessels [99, 101]. The role of advanced endoscopic techniques such as immersion endoscopy [102], NBI [103], iSCAN [104], and confocal endomicroscopy [105] is still unclear [106]. Capsule endoscopy may be useful for patients who refuse endoscopy and in complicated CD [107].
Histology is crucial for the diagnosis of CD in adults, and the way endoscopic biopsies are collected determines their accuracy. At least 4 duodenal biopsies should be collected, since the distribution of lesions is discontinuous [82]. Furthermore, 1 or 2 biopsies should be collected from the duodenal bulb (from the 9 or 12 o’clock position), since it increases (by almost 10%) the sensitivity in adults [108]. Bulb biopsies should be interpreted carefully and sent separately from more distal duodenal samples. Indeed, less than half of all bulb biopsies are high quality; their quality is hampered by the presence of Brunner glands and lymphoid tissue, peptic duodenitis, and gastric metaplasia. Furthermore, villi are smaller in the bulb and can be misinterpreted as atrophic [109, 110]. Finally, biopsies should be collected one-bite (rather than double-bite) at each pass of the forceps, since this decreases the risk of losing specimens, increases (3-fold) the chance of good orientation, and minimizes the risk of tangential biopsies that overestimate mucosal atrophy [111, 112]. Duodenal biopsies should be performed under a gluten-containing diet to avoid false negatives [90].
Histopathological findings characteristic of CD are intraepithelial lymphocytosis (≥25 IEL/100 enterocytes when sections are 3-4 um), crypt hyperplasia, and villous atrophy [90, 113]. A normal histology presents a gradient of IEL (more numerous at the base of the villi and decreasing toward the tip), and the villous:crypt ratio is 3-5:1 [114]. Histological abnormalities are classified according to the Marsh scale, modified by Oberhuber [113] as shown in Table 3.

Children tend to present, more frequently, mucosal atrophy (Marsh 3 lesions) on duodenal biopsies, whereas it is common to diagnose adults without villous atrophy and an inflammatory pattern with IEL (Marsh 1 or 2) [115].
A positive serology associated with a compatible histology (Marsh 2-3) confirms the CD diagnosis [73, 116].
Even though genetic testing is not required for the diagnosis of CD, it may be important to exclude CD, since the absence of HLA-DQ2/8 has a negative predictive value of virtually 100% [117]. This is particularly useful in seronegative patients, patients on a GFD who are unable/unwilling to undergo a gluten challenge, or those who refuse endoscopy [73, 92, 93, 118].
Research on CD is moving toward an endoscopy-free diagnosis of CD. Keeping that in mind, a study found a positive predictive value of 100% with the triple combination of a positive genetic test, positive anti-EMA, and over 10-fold increased anti-tTG titers;. In fact, recent European guidelines state that, in children, a duodenal biopsy may not be required if anti-tTG IgA is more than 10 times the upper limit of normal and anti-EMA IgA is positive in a second blood sample. In such cases, a serology-based diagnosis can be established even in the absence of symptoms and without genetic testing. On the other hand, when anti-tTG IgA titles are positive but low, duodenal biopsy should always be performed [84].
Furthermore, several noninvasive markers, such as intestinal fatty acid binding protein (I-FABP), a marker of enterocyte injury, are currently being evaluated. Patients with CD present higher serum levels compared to controls, which correlate with mucosal atrophy. I-FABP levels normalize after a GFD in 80% of children but not in adults [120]. Some studies also suggest a role of I-FABP in assessing adherence to a GFD and accidental gluten ingestion [121].
Another promising technique is flow cytometry that recognizes blood CD4+ T cells that bind to HLA-DQ-gluten tetramers. Preliminar studies have shown a very good accuracy in differenciating CD patients from controls, even on a GFD [122, 123].
Diagnostic Challenges
Seronegative-CD refers to a compatible histology and HLA, with a negative serology, and corresponds to 2% of CD patients [124, 125]. To confirm seronegative CD, the histology must improve after GFD. However, GFD is advised only after excluding other diagnoses, since seronegative CD accounts for <30% of seronegative villous atrophy or epithelial lymphocytosis [90, 126, 127]. As such, these should be excluded: autoimmune enteropathy (anti-enterocyte antibody positive), common variable immunodeficiency, Crohn disease, eosinophilic gastroenteritis, infectious diseases (Whipple disease, G.lamblia, tuberculosis, HIV-associated enteropathy, and tropical sprue), bacterial overgrowth, lymphoproliferative diseases, and drug-associated enteropathy. The drugs most frequently implicated are nonsteroidal anti-inflammatory drugs, immunosuppressors (azathioprine, mycophenolate mofetil, and methotrexate), and angiotensin receptor antagonists, in particular olmesartan, which is responsible for one fifth of seronegative duodenal-atrophy cases in the USA [116, 128-130]. In CD, IEL is composed solely of CD8+ T cells, whereas in non-CD villous atrophy IEL has mixed CD8+ and CD4+ T cells. CD-associated lymphocytosis is also suggested when over 5% of T-cell receptors in IEL are γ/δ and when the base-tip decrescendo gradient is lost [90, 114, 131]. Importantly, non-CD mucosal atrophy reverts spontaneously, without GFD, in over two thirds of patients [126].
Compared to seropositive patients, seronegative CD patients tend to be older (age 49 vs. 36 year) but they present more frequently the classic phenotype [125]. The physiopathology of seronegative CD is not yet clear, but some studies have suggested a high antibody-antigen binding affinity entrapping antibodies in the lamina propria away from the bloodstream. Accordingly, detection of tTG-anti-tTG immunocomplexes in the mucosa can help to identify these patients [132]. Other explanations for seronegative CD are immune system immaturity [133], selective IgA deficiency, a diet poor in gluten, treatment with immunossupressors, and refractory long-term CD [126].
Potential CD (PCD) refers to seropositive patients with a normal duodenal mucosa (Marsh 0) or intraepithelial lymphocytosis (Marsh 1) without crypt hyperplasia or villous atrophy. PCD accounts for 10% of CD patients [93]. Whereas over 80% of children are asymptomatic [134, 135], the majority of adults (79%) with PCD are symptomatic, mostly with a nonclassic phenotype [136]. Symptomatic PCD patients should be kept on a GFD, since it results in clinical improvement. The management of asymptomatic PCD patients is less straightforward, since the progression rate to overt CD is low, i.e., 13% in 10 years [137]. Asymptomatic PCD patients may maintain a gluten-containing diet, with evaluation every 6 months for symptoms and serology and a duodenal biopsy every 2 years if there is a persistently positive serology [136].
Patients on a GFD prior to CD diagnosis may be tested for HLA since the absence of HLA-DQ2/8 excludes CD. In the presence of HLA-DQ2/8, patients should repeat the diagnostic workup after a gluten challenge [138]. Traditionally, a gluten challenge consists of consumption of 7.5 g/day of gluten for 6-8 weeks; however, 3 g/day of gluten (equivalent to 2 slices of bread) is as effective. For patients who cannot tolerate a long gluten challenge, recent studies suggest that 2 weeks may be enough. One caveat is that, whereas histology can be performed immediately after the challenge, serology must be postponed 2 more weeks after the 2-week challenge. This approach allows a correct diagnosis in more than 75% of CD patients [138].
Non-celiac gluten sensitivity (NCGS) is a functional disorder that must be differentiated from CD. NCGS is 6 times more prevalent than CD [139], and it is more frequent in females in their second or third decade [140]. Clinical manifestations are elicited by gluten ingestion and they are similar to CD [141]; however, the kinetics between gluten ingestion and symptoms is much faster; intestinal or extra-intestinal symptoms develop and resolve hours to days after gluten ingestion or eviction [142, 143]. Furthermore, NCGS is not associated with autoimmune disorders. NCGS diagnosis is clinical with demonstration of symptoms resolution with a GFD, and recurrence after rechallenge, and requiring exclusion of CD and wheat allergy [141, 144]. The pathogenesis is unknown, though it is probably multifactorial, resulting from an interplay between the environment (other components of wheat), intestinal barrier dysfunction, gut dysbiota, and diregulated innate imune responses [140].
Management
A lifelong GFD is the only proven treatment and is recommended for classical, nonclassical, seronegative CD, symptomatic PCD, and HD or gluten ataxia [93]. GFD is not recommended for asymptomatic adults with PCD, since only a minority of these patients will develop villus atrophy [136].
A GFD consists of a strict elimination of wheat (and its gluten-containing derivatives bulgur, couscous, and seitan) [89], rye, and barley [145]. Elimination of oats is not so straightforward. In fact, oats contain avenin, a peptid related to gluten that may elicit similar immune reactions. Oats can also induce symptoms by an increase in fiber content. Hence, the oat intake should not exceed 50-60 g/day and patients should be clinically and serologically monitored. Oats should be avoided in severe disease [146].
The tolerable amount of gluten is variable, but as little as 1/100th of a slice of bread (around 50 mg of gluten) is sufficient to induce mucosal atrophy. Gluten-free is defined as <20 ppm of gluten (around 6 mg/day)[147, 148]. Patients should be aware of nondietary potential sources of gluten contamination such as tooth paste and lipstick.
Gastrointestinal symptoms improve after 1 month and usually disappear after 6 months on a GFD [149]. Most patients become seronegative after 6 months on a GFD and only 17% remain seropositive after 1 year [150, 151], suggesting gluten contamination [73]. Anti-tTG IgA is the preferred serological test to monitor GFD adherence. Histological normalization takes longer, particularly in adults, in whom it takes 2-5 years [152]. Only 66% of patients on a GFD achieve a total histological recovery after 1 year, in contrast with the expected recovery in 95% of children [153].
A GFD can also improve extraintestinal manifestations and CD-related conditions. However, some manifestations such as enamel defects and osteopenia may be irreversible or just partially corrected [66, 154].
The GFD should be lifelong, even if the patient acquires a clinical tolerance to gluten. Although 20% of patients maintain histological remission after gluten reintroduction, IEL and a positive serology tend to remain, and those patients are at an increased risk for extraintestinal manifestations and late relapse [155]. Importantly, strict adherence to a GFD is low, i.e., 17-48% [156, 157], and mortality seems to increase 5-fold in patients who do not comply with a GFD [158].
Alternative therapies are under investigation but none has shown sufficient efficacy yet to enter clinical practice. Investigational drugs include genetically modified less immunogenic wheat strains, prolyl endopeptidases, nonabsorbable polymers with a high affinity for gliadin, drugs that act on intestinal permeability gluten deamination, and HLA inhibitors, among others [159].
Patients should be monitored at 6 months and then yearly for GFD adherence, symptoms, serology, a micronutrient deficiency, and associated conditions. Laboratorial tests should include anti-tTG IgA, a full blood count, iron, folic acid, vitamin B12, calcium, vitamin D, thyroid function, and anti-thyroid antibodies [26, 152]. Follow-up endoscopy is advised for persistent or relapsing symptoms despite a GFD [82]. Osteodensitometry should be assessed every 1-2 years [89]. Lastly, vaccination against pneumococci, Haemophilus influenza, and meningococci are strongly recommended [73].
About 1.5% of CD patients progress to refractory CD (RCD), defined as persistence of clinical malabsorption and villous atrophy, after 1 year on GFD, after exclusion of other causes for villous atrophy or malignancy [116]. The main cause of persistent villous atrophy is inadvertent gluten ingestion. Other conditions, i.e., lactose intolerance, irritable bowel syndrome, small bowel bacterial overgrowth, pancreatic insufficiency, and microscopic colitis, should be excluded [83].
RCD is subclassified into types I and II, according to phenotype and clonality of IEL. In type I RCD, IEL are phenotypically normal with polyclonality of the T-cell receptor, whereas in typeII T cells are aberrant, lacking surface CD8 and CD3 expression while expressing intracytoplasmatic CD3, and presenting a monoclonal receptor rearrangement. The distinction of these 2 entities is crucial because the treatment and prognosis are different [89, 160, 161]. Type I RCD usually responds to steroids and budesonide or immunomodulators such as azathioprine. Type II RCD is more aggressive and it is associated with ulcerative jejunoileitis, severe malabsorption, a high risk of progression to enteropathy-associated T-cell lymphoma (EATL) (50%, in 5-10 years) [154, 162], and a 5-year survival rate of 44-58% [154, 160]. Type II RCD does not respond to steroids, should not be treated with azathioprine because of concerns of an increase in the risk for EATL, and may require treatment with cladribine or an autologous/allogenic bone marrow transplant. Targeting of IL-15 is a promising therapeutic strategy [163].
Patients with CD, especially long-standing and untreated patients, present a higher risk for EATL and small intestine adenocarcinoma compared to the general population. The 5-year survival rate for EATL is 11%. The risk of developing other malignancies is still an unanswered topic [89, 164-166].
CD patients seem to have a 20% increase in mortality, particularly in those diagnosed as young adults and in the first 2 years after the diagnosis. Of note, increased mortality in CD patients seems to occur even 10 years after the diagnosis [167]. The mortality rate is probably influenced not only by the age at diagnosis but also by the severity of the presentation, the adherence to a GFD, the ammount of gluten intake, and associated conditions [168].
Specific mortality increases in lymphoproliferative disorders, with a 2-fold increase particularly in women over 50 years of age and in the 2 years following the diagnosis. In fact, in a Finnish study on 12,803 CD patients followed for 7 years, 55% of CD patients died from T-cell lymphoma, compared to 3% of the reference population [169]. Globally, however, no increased risk for cancer-related mortality has been proven [166]. Importantly, CD patients seem to have a 5-fold increased risk of dying from infections, particularly1 sepsis [169].
Conclusions
CD is still an underdiagnosed entity that poses a diagnostic challenge despite having been described, for the first time, almost 2 thousand years ago. It presents a wide range of unspecific signs and symptoms, both gastrointestinal and extraintestinal. Adults tend to be paucisymptomatic, presenting nonclassical symptoms which can also occur in children. As such, even though population-based screening is not recommended, physicians should use an active case-finding strategy with a low threshold for screening.
Diagnosis requires highly accurate serological tests and a compatible duodenal histopathology, although in children duodenal biopsy may be avoided if the anti-tTG is higher than 10 times the upper limit of normal and anti-EMA is positive in a second blood sample. The presence of HLA-DQ2/8 is mandatory for the development of CD, being particularly helpful in excluding CD. Typical histology findings such as villous atrophy and crypt hyperplasia are unspecific and other diseases, such as Crohn disease, infections, and drug-induced duodenitis, must be excluded.
A lifelong GFD is the only treatment with proven efficacy; however, adherence to this diet is very low. As such, a GFD must be emphasized and monitored regularly. CD patients seem to have an increased mortality, most likely if left untreated.
Current and future research on CD should address endoscopy-free diagnostic algorithms, easier monitoring of dietary gluten contamination, and alternative nondietary therapeutic strategies.

