











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
The most frequent mesenchymal neoplasms affecting the gastrointestinal (GI) tract are GI stromal tumors (GIST). GIST are most often located in the stomach (60-70%), followed by the small bowel (20-35%), the colon and rectum (5%), and the esophagus (<5%) [1, 2]. Although the majority of GIST are sporadic, approximately 5% of patients have 1 of several familial autosomal dominant syndromes, such as primary familial GIST syndrome, neurofibromatosis type 1 (NF1), or Carney-Stratakis syndrome ]3[.
These tumors are identified mainly by expression of the KIT protein (95%), and they frequently harbor activating mutations in the KIT or platelet-derived growth factor-α PDGFRA receptor genes [1]. The most prominent diagnostic marker of GIST is overexpression of the receptor tyrosine kinase KIT (CD117) (90%), which is easily recognized by positive immunohistochemical staining [1]. Almost 75% of GIST are composed of spindle cells, whereas the remaining GIST consist of epithelioid cells [1].
GIST are more frequently associated with GI bleeding and acute abdomen due to tumor ulceration and their large growth, respectively. Nevertheless, patients could be asymptomatic and GIST can be incidentally detected during endoscopic study or cross-sectional imaging, or patients could only report nonspecific symptoms (e.g., early satiety and bloating) [3].
Multiple procedures are commonly used to investigate small bowel tumors, although lesions presented as submucosal tumors may be detected more readily by capsule endoscopy [2, 4].
Case Report
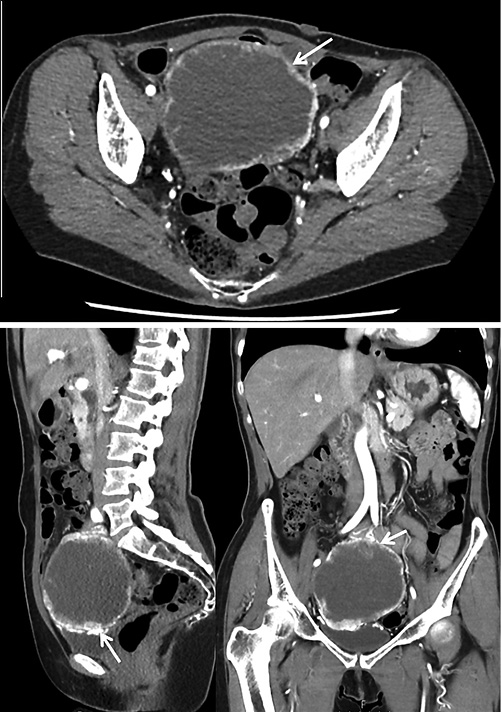
A 58-year-old woman presented to the emergency department with a 5-day history of melena. Her medical history included an inflammatory ileocolonic Crohn disease (CD) (Montreal classification: A2L3B1) and NF1, without current medication. Ileocolonoscopy and magnetic resonance enterography performed 3 and 4 years ago, respectively, were unremarkable, and she was in clinical CD remission. An abdominal examination did not reveal any pain or abnormal mass. A laboratory study revealed normocytic normochromic anemia (hemoglobin, 8 g/dL; mean corpuscular volume, 82.6 fL; and mean corpuscular hemoglobin concentration, 32.5 g/dL). She underwent an esophagogastroduodenoscopy, the results of which were normal. Taking into account her CD history, a panenteric capsule endoscopy was performed. Capsule endoscopy did not detect the presence of active disease, although it demonstrated an ulcerated subepithelial lesion with bleeding stigmata presumably in the proximal jejunum (Fig. 1 a-b). Anterograde single-balloon enteroscopy confirmed a 4-cm ulcerated protruding lesion, suggestive of a GIST, and a tattoo was placed proximally (Fig. 1 c-d). Computed tomography showed a heterogeneous enhanced small bowel exophytic lesion of around 9 cm, suggestive of a GIST, with intimate contact with the bladder, the appendix, and the sigmoid colon but without evident invasion of those structures (Fig. 2). The patient underwent laparoscopy and small-bowel resection with end-to-end anastomosis. During surgery, tumor rupture was verified. Pathology confirmed a GIST of the epithelioid cell subtype, with a mitotic rate of 2/5 mm2, without KIT or PDGFRA mutations. Margins were free of tumor, and the TNM staging was pT3. By immunohistochemistry, cells were positive for CD34, CD117, and DOG-1. AT the time of writing, the patient was on imatinib and fluorodeoxyglucose positron emission tomography performed 3 months after surgery revealed no evidence of relapse.
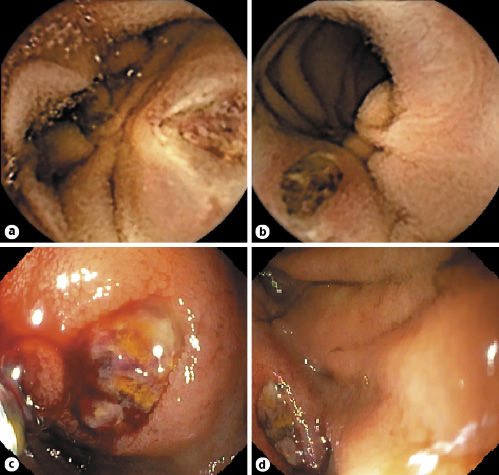
Fig. 1 Small bowel findings of a 4-cm ulcerated subepithelial lesion with bleeding stigmata suggestive of a GIST. a, b Capsule endoscopy images. c, d Single-balloon enteroscopy images.
Discussion
GIST arising in the GI tract are typically found as a subepithelial lesion, and as they grow this could lead to epithelial ulceration which could ultimately manifest as GI bleeding [2, 5, 6]. Individuals, like our patient, with NF1 have a significantly increased risk of GIST (5-25%) especially in the small bowel (>70%), and, unlike the sporadic type, these patients usually do not carry somatic mutations in the KIT or PDGFRA genes [7]. Curiously, overexpression of KIT may be present even in the absence of KIT mutations, especially in the setting of NF1, as noticed in this patient’s tumor with CD117 staining [7].
GIST have potential for malignant behavior, and some prognostic factors were already evaluated, such as tumor size (>5 cm), mitotic activity (>5 mitoses per 50 HPF), tumor location (small bowel or rectal), depth invasion, grade differentiation, tumor rupture, and presence of KIT mutation. Furthermore, some other tumor characteristics on CT or endoscopic ultrasound, such as lobulated morphology, heterogeneous enhancement, mesenteric fat infiltration, ulceration, regional lymphadenopathy, or an exophytic growth pattern, are considered to be more susceptible to metastasis [8]. In patients with a risk of relapse, adjuvant treatment with imatinib at 400 mg for 3 years is advisable [3]. Even though our patient presented some high risk features, such as the presence of a small-bowel large tumor with heterogeneous enhancement, exophytic growth, and tumor rupture, since NF1-GIST oncogenesis is attributed to activation of the RAS/RAF/MAP kinase signaling pathway, these tumors rarely respond to imatinib and no standard drug therapy for relapsed tumors has been established [7, 9]. In the present case, the authors reluctantly administered imatinib as first-line therapy since another tyrosine kinase inhibitor was not available.
Our report emphasizes the importance of small-bowel investigation in this patient with 2 simultaneous important chronic entities, i.e., CD and NF1, who presented an overt bleeding episode. Although the patient’s clinical presentation could have misled us to suspect of a CD flare, the association between NF1 and GIST motivated a timely capsule endoscopic evaluation, which prevented a delay in the subsequent management, i.e., a surgical approach. As small bowel GIST are considerably incident in patients with NF1, it is questionable whether these patients should be screened with capsule endoscopy. As has been previously recognized [10], there is not yet sufficient evidence to support this type of screening program in this group of patients.

