











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
Permalink
Introduction
Acute liver injury (ALI) is characterized by an acute abnormality of liver blood tests in patients without underlying chronic liver disease, who develop liver-associated coagulopathy, but as opposed to acute liver failure (ALF), without any change in level of consciousness [1]. Viral infections and hepatotoxic drugs are the most common causes of severe ALI [2]; however, these features can be seen in various systemic disease processes [1].
Hemophagocytic lymphohistiocytosis (HLH) is a rare, potentially life-threatening hyperinflammatory syndrome associated with signs and symptoms consequent to extreme and ineffective immune activation [3-5]. HLH may be due to primary or secondary causes. Secondary causes result from acquired immune dysfunction in response to infections, malignancies, autoimmune diseases, and toxics; few case reports described HLH following administration of vaccines [3].
The clinical presentation is heterogeneous and non-specific and may even course with multiorgan failure [4]. Cardinal features are continuous high fever, cytopenias, and hepatosplenomegaly [1]. Liver involvement is com-mon; however, presentation with severe ALI/ALF is rare [3], which can lead to delays in diagnosis and therapy initiation, with consequent fatal outcome. Since there is no single diagnostic test, it is essential high index of suspicion and the application of the HLH-2004 criteria [3].
Case Presentation
A 65-year-old Caucasian man presented with persistent high fever (>39°C), myalgias, and jaundice 1 week after the first dose of COVID-19 vaccine. His medical history was remarkable for a low-risk chronic lymphocytic leukemia (CLL) (Rai 0, Binet A) with no symptoms under active monitoring, rheumatoid arthritis previously on prednisolone (10 mg/day), and traumatic splenectomy. There was no relevant epidemiological background.
Upon admission, on physical examination the patient was hemodynamically stable, febrile (40°C), frankly jaundiced and a non-painful hepatomegaly was detected; the remaining examination was unremarkable, with no signs of encephalopathy, ascites, chronic liver disease, active arthritis, mucocutaneous bleeding, cutaneous purpura, or enlarged lymph node. Initial blood investigations revealed bicytopenia, severe liver injury, and increased INR with normal remaining coagulation (shown in Table 1). Periph eral blood smear showed predominance of mature lymphocytes. Computed tomography chest-abdomen-pelvis excluded signs of malignancy, enlarged lymph nodes, chronic liver disease, or biliary obstruction.
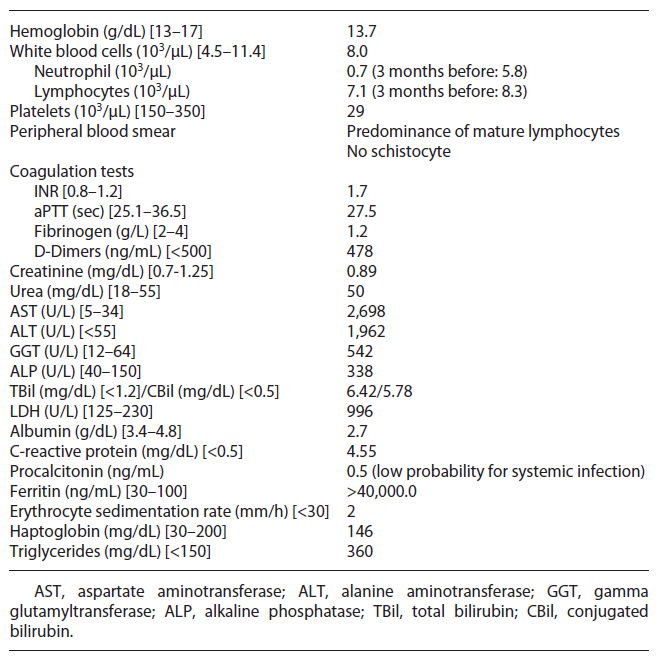
Initial septic workup (urine examination and imaging tests) ruled out infection; however pending the results of cultures, the patient was on antibiotic therapy. After 72 h, he remained febrile and developed organ failure: cardiovascular (systolic arterial pressure <90 mm Hg requiring inotropic support) and respiratory (PaO2/FiO2: 200-300 mm Hg requiring supplementary O2), so he was admitted to the intermediate care unit. Cultures have proven sterile, and extensive workup for liver disease was normal (shown in Table 2). Further workup revealed high ferritin, LDH, and triglycerides and low fibrinogen (shown in Table 1).
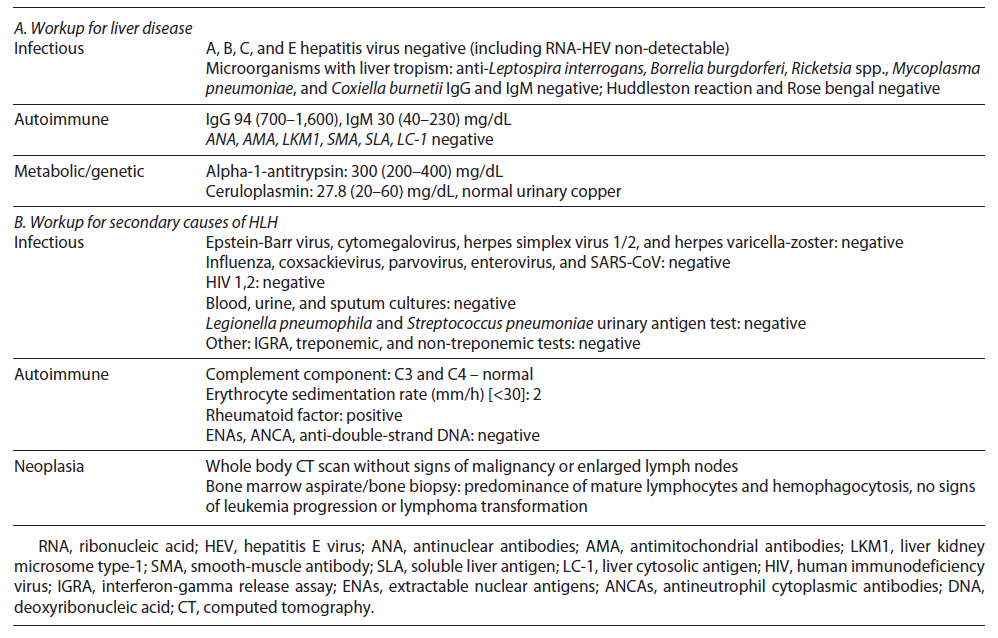
After excluding malignancy (normal computed tomography), infections (negative cultures and normal wide virus panel), and rheumatic disorder flare (no arthritis and normal complement, autoimmunity, and erythrocyte sediment rate), we considered HLH diagnosis attending to the cytopenias, hyperferritinemia, and persistent fever. An HLH score of 277 supported the diagnosis. Attending to organ failure, while we were waiting for the results of the bone marrow aspirate, we started dexamethasone (10 mg/m2 daily) and first dosage of etoposide (150 mg/m2). Meanwhile, the bone marrow aspirate showed hemophagocytosis, predominance of mature clonal lymphocytes, and no signs of progression nor transformation to aggressive lymphoma. The patient continued treatment with etoposide (150 mg/m2 biweekly for 2 weeks, then weekly) and dexamethasone (10 mg/m2 daily for 2 weeks with progressive tapering).
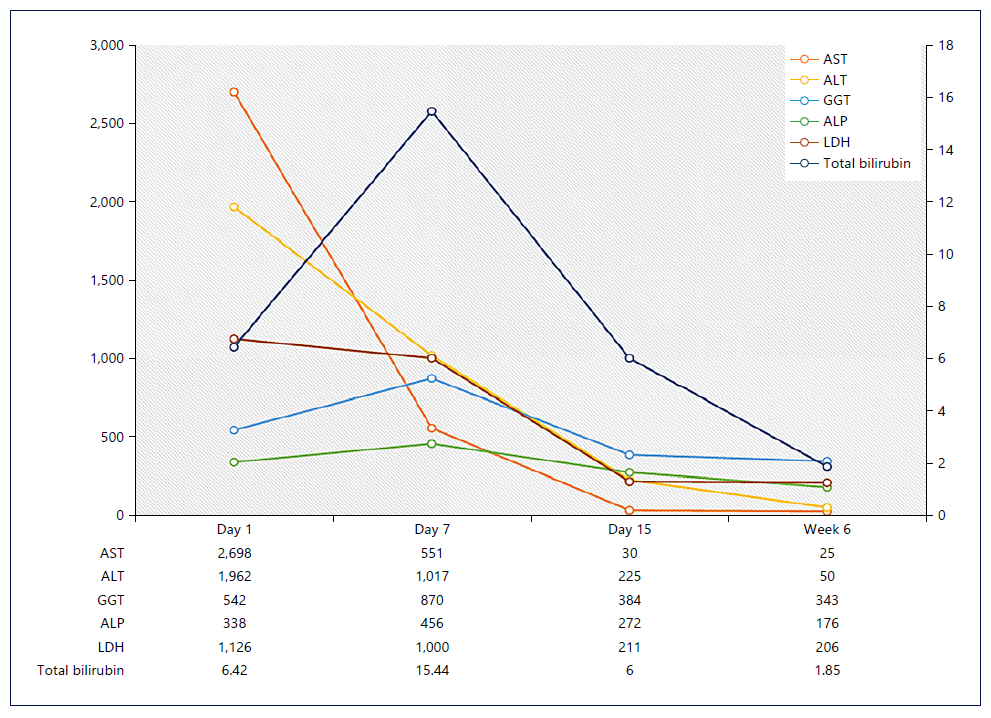
Rapid clinical (apyrexia and reversal of all organ failures) and liver improvement was observed (shown in Fig. 1), as well as neutrophil count and INR normalization. However, after 6 weeks of therapy, the patient developed febrile neutropenia related to immunomodulatory therapy, so we discontinued etoposide and started granulocyte colony-stimulating factor and broad-spectrum antibiotic therapy. The patient remained neutropenic and acquired a healthcare-associated pneumonia requiring intensive care admission, there was a progressive worsening, and he died 8 weeks after admission.
Discussion
HLH results from uncontrolled activation of macrophages, natural killer cells, and T cells due to enhanced antigen presentation [3, 4]. This activation produces massive secretion of proinflammatory cytokines, a socalled cytokine storm, that directly contributes to end-organ damage and rapidly progressive multiorgan dysfunction [4].
The etiology of HLH is classified into primary (genetic) and secondary (reactive). In children, congenital defects in cytotoxic T cell and natural killer cell function and inflammasome dysregulation are described. Secondary causes result from acquired immune dysfunction in response to infections, malignancies, autoimmune diseases, or other causes (e.g., drugs, vaccination, organ/stem cell transplantation) [3, 6]. The underlying cause cannot be identified in 20% of cases [6, 7].
CLL is a monoclonal lymphoproliferative disease that results from the proliferation and accumulation of morphologically mature but immunologically dysfunctional B-cell lymphocytes [8]. HLH in the context of CLL has rarely been reported, mostly due to chemotherapy and CLL progression/transformation [9]. We emphasize that our patient had no evidence of progression of CLL or transformation into a more aggressive lymphoma. In CLL, there is a continuous crosstalk between dysfunc tional B and T lymphocytes [10, 11]. Disturbances in apoptosis of T cells, altered patterns of surface molecules of T cells, and unbalanced cytokine environment (IL-10, IL-6, IL-4) were described in CLL [10], which might contribute to uncontrolled activation of the immune system. Secondary HLH may be induced by autoimmune disorders, known as macrophage activation syndrome. Although our patient had rheumatoid arthritis, there were no arthritis nor analytical alteration suggesting flare.
Few case reports have described HLH following administration of vaccines [7, 12-14] including after COVID-19 vaccination [15-17], mostly in children with in-herited variants of HLH-associated genes or immunosuppressed adults. We postulated that the uncontrolled activation of the immune system resulted from an immune response triggered by vaccination (time frame) and an underlying immune defect (immunosuppression, malignancy). Dramatic proinflammatory responses have been identified in some individuals following immunization [18].
The clinical presentation of HLH is mostly non-specific with an acute or subacute course [3]. The liver is one of the most affected organs. ALT and AST elevations are seen in ∼85% of adult, mostly mild, and half of patients have hyperbilirubinemia [6, 7]. ALI/ALF as presentation is rare and usually occurs with concomitant organ failure [19]. Liver histopathological features include sinusoidal dilatation and hepatocellular necrosis [20]. Underlying liver disease, infiltration of activated histiocytes and over-production of cytokines are the putative mechanisms of liver injury [21].
Pulmonary (42%), cutaneous (25%), and neurological (25%) involvement are also frequent [3, 22]. To make the diagnosis of HLH, the patient should meet five of the eight diagnostic HLH-2004 criteria (shown in Table 3) [23].

Cytopenias are the key laboratory markers of HLH. Hyperferritinemia is the finding that most often (90%) leads to suspicion of LHH [3, 23]. Hypertriglyceridemia or hypofibrinogenemia is easily determined. Although determination of soluble CD25 is helpful for diagnosis, it is rarely available [23]. Finally, the demonstration of hemophagocytosis in bone marrow is helpful for diagnosis, being also mandatory to exclude underlying malignant disorders [23]. In our patient, beyond the signs of hemophagocytosis in the bone marrow, there was no evidence of CLL progression nor transformation to aggressive lymphoma, so five criteria were met.
The HScore [24], which includes clinical and laboratory parameters, supports the diagnosis. Our patient predicted HLH with 99% of probability, so we started therapy immediately, given the presence of organ failure. The diagnosis of severe liver dysfunction induced by HLH is challenging, particularly in the early phase of the disease, as the presentation is non-specific, making it difficult to distinguish it other causes of ALI [3].
The prognosis of adult HLH is poor, with mortality rates ranging from 41 to 75% [3, 6, 19]. A delayed diagnosis is the limiting step toward a successful outcome, including hyperinflammation control (corticosteroids and etoposide) and treatment of the underlying cause, based on HLH-2004 protocols [3]. Early suspicion supported by a high HScore allowed the timely institution of successful treatment. The patient’s outcome was conse-quence of neutropenia and respiratory infection. Some authors recommend prophylactic antibiotic and antifungal therapy and granulocyte colony-stimulating factor in patients treated with T-cell depleting therapy [25], which was not started as early as desirable in our patient. Additionally, there is growing evidence of polyvalent immunoglobulin in HLH treatment [23], specially in our patient, as CLL is associated with lower humoral immunity and hypogammaglobulinemia.
HLH requires a high index of suspicion and should be considered in patients with unknown cause of severe liver dysfunction accompanied by sudden unexplained on set of systemic inflammatory response and multiple organ involvement. We alert to the need to monitor suspicious symptoms after vaccination (e.g., high fever) in patients with underlying pathology. The diagnosis of HLH should not be based on the fulfilment of criteria alone, and it is of utmost importance that an experienced hematologist judges the clinical aspects and weighs up the risks and benefits of treatment.

