











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkMechanism of action of the JAK-STAT pathway
The Janus Kinase (JAK) and signal transducer and activator of transcription (STAT) pathway is a ubiquitous intracellular signaling network that is involved in the signal transduction of numerous dermatologically relevant cytokines1,2. JAK family is composed of four members: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). The function of JAK proteins is associated with the type of cytokine that binds the receptors and they play a key role in cell growth, development and differentiation. They are especially found in immune and hematopoietic cells, such as lymphocytes, natural killer cells, and mast cells, but cytokine signaling is also important for the biology of nonimmune cells such as keratinocytes, fibroblasts, osteoblasts, synoviocytes, or endothelial cells. JAK1, JAK2, and TYK2 are involved in cell growth processes in different cell types, while JAK3 is critical to hematopoiesis3. Different JAKs are associated with specific cytokine receptors and influence different aspects of immune cell development and function. These proteins modulate the inflammatory process by activating intracytoplasmic transcription factors called STAT. STAT family influences DNA transcription and plays an important role in regulating gene expression, cell differentiation, proliferation, survival and apoptosis. This family is composed of seven proteins (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6); phosphorylated STATs dissociate from the receptor, translocate to the cell nucleus and regulate transcription either positively or negatively of thousands of different genes (Figure 1).
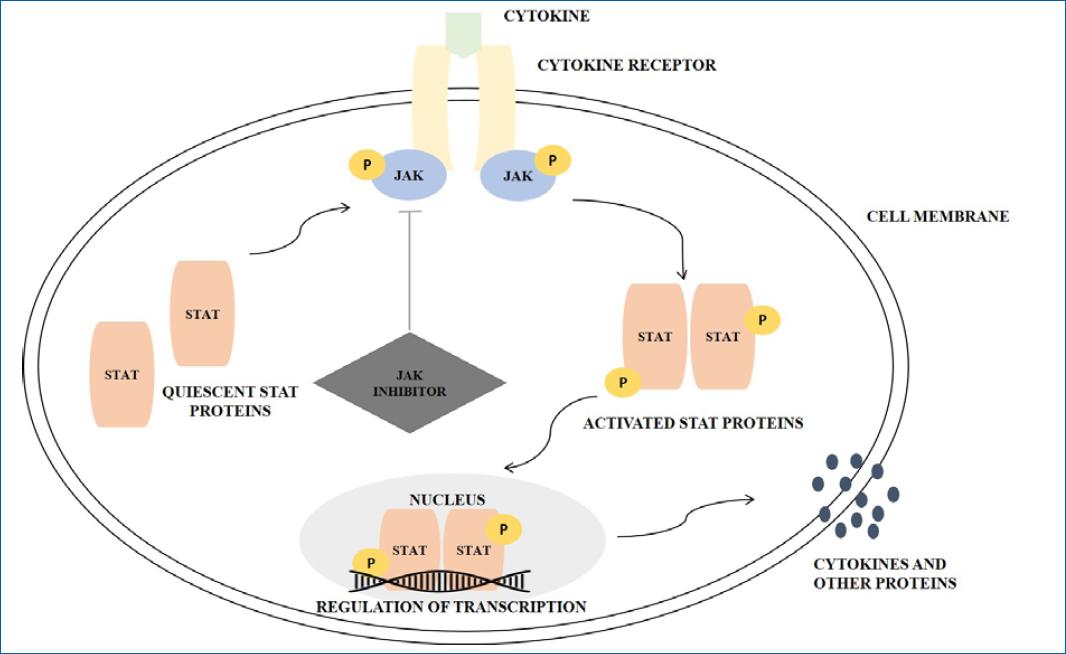
Figure 1 Schematic presentation of the JAK/STAT pathway and the role of JAK inhibition drugs. The primary function of the protein kinases is to transfer phosphate groups from adenosine triphosphate or guanosine triphosphate to the hydroxyl groups of amino acids of the protein targets. Numerous group of cytokines, such as IL-2, IL-6, IL-12, IL-21, IL-22, IL-23, and INF-Υ interacts with type I and II cytokine receptors. Both of these receptor types lack intrinsic enzyme activity and rely on JAKs for signal transduction. After binding, recruited JAKs to initiate a signaling pathway: cytokine receptors of type I and II undergo oligomerization leading to the recruitment of JAKs, which autophosphorylate tyrosine residues. Then, STAT proteins are recruited and bind to the phosphorylated residues leading to activation mediated by phosphorylation by JAKs. Successively, the activated STAT proteins undergo dimerization enabling their translocation to the nucleus and, consequently the modulation of gene expression (adapted from Solimani et al.)2.
Thus, selective JAK inhibitors can simultaneously block the function of multiple cytokines and, consequently, the transcription of genes responsible for inflammation and the control of innate and adaptative immunity. Most cytokine receptors use a combination of JAKs for their activity, enabling the idea of targeting single JAKs. JAK inhibitors are divided into two generations, the first generation blocks more than one type of JAK, while the second generation is more specific and blocks only one type of JAK and consequently has fewer side effects3. The first-generation JAK inhibitors include tofacitinib, ruxolitinib (RUX), baricitinib, and oclacitinib, all of which are approved for use in humans, except oclacitinib4. Abrocitinib and upadacitinib are commonly referred to as selective JAK1 inhibitors2,4.
Rational for the use of JAK-inhibitors in dermatology
The perpetuation of inflammation in diseased skin strongly relies on the interaction between cytokines, immune, and tissue cells propagating distinct inflammatory cascades. Differently from biologics that target cytokines by intravenous or subcutaneous injection, JAK inhibitors target cytokine signaling by either oral or topical administration. The latter way of application may minimize the risk of side effects. Topical JAK inhibitors do not bear the risk of skin atrophy or telangiectasia, as observed with long courses of topical corticosteroids2. There is growing interest in the potential use of these drugs in many dermatological diseases, such as alopecia areata (AA), vitiligo, and atopic dermatitis (AD). The cytokines involved in each of these diseases differ; however, in all three, the effects of cytokine binding are mediated through the JAK pathway, providing a rationale for JAK inhibition. Although there are many JAK inhibition under study, only some are approved for the treatment of dermatological diseases (Table 1).
Table 1 Summary of FDA and EMA approval of JAK inhibitor drugs in dermatologic diseases and psoriatic arthritis
| Main target | EMA approval | FDA approval | Route of administration | |
|---|---|---|---|---|
| Abrocitinib (CIBINQO®)8,74 | JAK1 | Adults with moderate-to-severe AD who are candidates for systemic therapy | Adults with refractory, moderate-to-severe ADª | Oral |
| Baricitinib (OLUMIANT®)9,19 | JAK1, JAK2 | Adults with moderate-to-severe AD who are candidates for systemic therapy; adult patients with severe AA | Adult patients with severe AA | Oral |
| Tofacitinib (XELJANZ®)75,76 | Predominantly a JAK1/3 inhibitor with functional selectively over JAK2 | In combination with MTX for the treatment of active psoriatic arthritis (PsA)b | Adult patients with active PsA with inadequate response or intolerance to methotrexate or other DMARDs | Oral |
| Upadacitinib (RINVOQ®)7,77 | JAK1 | Active PsA in adult patients who have responded inadequately to, or who are intolerant to one or more DMARDsd Treatment of moderate to severe AD in adults and adolescents 12 years and older who are candidates for systemic therapy | Adults with active PsA who have had an inadequate response or intolerance to one or more TNF blockers Adults and pediatric patients 12 years of age and older with refractory, moderate to severe ADc | Oral |
| Deucravacitinib (SOTYKTU®)60 | TYK2 | Under regulatory review for the treatment of moderate-to-severe plaque PSO | Adults with moderate-to-severe plaque PSO who are candidates for systemic therapy or phototherapy | Oral |
| Ruxolitinib (OPZELURA®)39 | JAK1, JAK2 | Decision agreeing on an investigation plan. Validation form EMA for a potential treatment for ≥ 12 years with nonsegmental vitiligo with facial involvement | Short-term/noncontinuous chronic treatment of mild to moderate AD in nonimmunocompromised ≥ 12 yearse Topical treatment of nonsegmental vitiligo in ≥ 12 years | Topic |
EMA: European Medical Agency; FDA: Food and Drug Administration; EU: European Union; AD: atopic dermatitis; AA: alopecia areata; DMARDs: disease-modifying antirheumatic drugs;
ª: whose disease is not adequately controlled with other systemic drug products, including biologics, or when the use of those therapies is inadvisable;
b: in adult patients who have had an inadequate response or who have been intolerant to a prior disease-modifying antirheumatic drug (DMARD) therapy;
c: whose disease is not adequately controlled with other systemic drug products;
d: may be used as monotherapy or in combination with methotrexate;
e: whose disease is not adequately controlled with topical prescription therapies.
Atopic dermatitis
Atopic dermatitis (AD) is a common chronic disease of the skin that has a great impact on a patient’s quality of life. The disease can manifest at any age, but its prevalence is higher in children and adolescents. Clinically AD is characterized by the presence of pruriginous eczematous lesions, typically on flexural sites. This heterogeneous condition has a complex pathophysiology; historically AD is thought to be T helper 2 (Th2) dominated disease, but there is growing evidence that the immunological environment of AD is not solely defined by Th2 cells and related cytokines but also by cytokines linked to other Th cells responses such as INF-Υ [(interferon-gamma) Th1], interleukin (IL)—17, IL-22 (Th17) and IL-332. Given the diversity of cytokines implicated in the inflammatory process of AD, there is growing interest in JAK inhibition, which could interfere with multiple cytokines simultaneously.
Various cytokines relevant to the pathophysiology of AD, including IL-4, IL-13, IL-22, IL-31, and thymic stromal lymphopoietin, activate JAK1 containing heterodimeric receptors, thereby mediating Th2 cell differentiation5. Additionally, chronic itch is also directly mediated by neuronal JAK1 signaling, and IL-22 drives epidermal hyperplasia via JAK1. Together these findings suggest the importance of JAK1 signaling in the pathogenesis of AD6. JAK2 forms homodimeric receptor complexes involved in hematopoiesis. Therefore, selective inhibition of JAK1 is a desirable target to modulate a broad range of cytokines involved in the pathophysiology of AD while avoiding the effects of JAK2 inhibition, such as neutropenia and anemia5. A growing body of literature has demonstrated that JAK inhibitors are safe and efficacious in multiple inflammatory skin conditions, including AD2,4.
Systemic JAK inhibitors
The first-generation inhibitor of JAK1/2, baricitinib, and JAK1 selective inhibitors, such as abrocitinib and upadacitinib were approved recently by the European Medical Agency (EMA) for the treatment of adult patients with moderate to severe AD. Upadacitinib has extended approval for children 12 years of age and older7-9. The approval of these systemic JAK inhibitors was a major milestone in the treatment of AD (table 1 and table 2). The safety, and efficacy with rapid relief of pruritus and clinical signs of AD explored by clinical trials (table 3) make these agents a welcome addition to the box of therapeutic options for managing AD. Currently, the monoclonal antibody dupilumab will be the main competitor. Each has its advantages, as some patients prefer a subcutaneous injection with no laboratory monitoring, whereas others may prefer the convenience of oral therapy4. There are no reported studies that directly compare JAK inhibitors with each other; however, a recently published network meta-analysis aimed to determine the comparative efficacy and safety of three common oral JAK inhibitors, including abrocitinib, baricitinib, and upadacitinib for moderate-to-severe AD. This network meta-analysis revealed that upadacitinib 30 mg was superior to all regimens and upadacitinib 15 mg was better than remaining regimens except for abrocitinib 200 mg in terms of Investigator’s Global Assessment scale (IGA) and Eczema Area and Severity Index (EASI) response. Moreover, abrocitinib 200 mg was superior to abrocitinib 100 mg, and baricitinib 1, 2, and 4 mg for clinical efficacy. However, upadacitinib 30 mg caused more treatment-emergent adverse effects (AE)10.
Table 2 Posology recommended for abrocitinib, baricitinib, and upadacitinib
| Variables | Route of administration | Posology |
|---|---|---|
| Abrocitinib (CIBINQO®)8 | Oral | The recommended starting dose is 200 mg once daily (100 mg once daily is recommended for patients ≥ 65 years of age) During treatment, the dose may be decreased or increased based on tolerability and efficacy. Maximum dose 200 mg daily |
| Baricitinib (OLUMIANT®)9 | Oral | The recommended dose of baricitinib is 4 mg once daily (A dose of 2 mg once daily is appropriate for patients: aged ≥ 75 years; history of chronic or recurrent infections; sustained control of disease activity with 4 mg once daily, and are eligible for dose tapering) |
| Upadacitinib (RINVOQ®)7 | Oral | ≥ 18 years: The recommended dose of upadacitinib is 15 mg or 30 mg once daily based on individual patient presentation (For patients ≥ 65 years of age, the recommended dose is 15 mg once daily. The lowest effective dose for maintenance should be considered) 12-17 years: The recommended dose of upadacitinib is 15 mg once daily for adolescents weighing at least 30 kg |
Table 3 Summary of efficacy and safety data of clinical trials of systemic JAK inhibitors approved by European Medicines Agency for the treatment of atopic dermatitis
| Variables | Authors | Clinical trial | Assessment methods | Results | Safety |
|---|---|---|---|---|---|
| Abrocitinib1 | Simpson et al.5 | Phase 3, randomized, multicentric, double-blind, placebo-controlled trial (JADE MONO-1) 387 patients (≥ 18 years) Three groups of patients: abrocitinib 100 mg ID (n = 156), abrocitinib 200 mg ID (n = 154), placebo (n = 77) | Coprimary endpoints: Investigator Global Assessment response: score 0 (clear) or score 1 (almost clear) ≥ 75% improvement in EASI (EASI-75) score from baseline | Investigator Global Assessment response and EASI-75 at week 12: abrocitinib 100 mg ID group: 24 and 40%; abrocitinib 200 mg ID group: 44 and 63%; placebo group: 8 and 12%, respectively | AE was reported in 69% of patients in abrocitinib 100 mg ID group, 78% of patients in abrocitinib 200 mg ID group, and 57% of patients in the placebo group. Most common AE in the abrocitinib 100 and 200 mg group: nausea (n = 14, n = 31, respectively) and nasopharyngitis (n = 23, n = 18, respectively) SAE treatment-related: one patient in abrocitinib 200 mg group developed inflammatory bowel disease; one patient in abrocitinib 100 mg group developed acute pancreatitis |
| Bieber et al.78 | Phase 3, randomized, multicentric, double-blind, placebo-controlled trial (COMPARE) – 838 patients (≥ 18 years) – four groups of patients: abrocitinib 100 mg ID + topicals (n = 238), abrocitinib 200 mg ID + topicals (n = 226), dupilumab 300 mg every other week after a loading dose of 600 mg + topicals (n = 243), placebo (n = 131) |
Coprimary endpoints: investigator Global Assessment response: score 0 (clear) or score 1 (almost clear) ≥ 75% improvement in EASI (EASI-75) score from baseline | Investigator Global Assessment response and EASI-75 at week 12: abrocitinib 100 mg ID group: 36.6 and 58.7%; abroctinib 200 mg ID group: 48.4% and 70.3%; dupilumab group: 36.5 and 58.1%; placebo group: 14.0 and 27.1%, respectively (all p < 0.001) | AE was reported in 50.8% of patients in abrocitinib 100 mg group, 61.9% of patients in the abroctinib 200 mg, 50.0% of patients in dupilumab group, and 53.4% of patients in the placebo group. The most common AE with abrocitinib was nausea, acne, nasopharyngitis, and headache SAE reported: 2.5% in abrocitinib 100 mg group, 0.9% in abrocitinib 200 mg group, 0.8% in the dupilumab group, and 3.8% in the placebo group | |
| Silverberg et al.79 | – Phase 3, parallel-group randomized, multicentric, double-blind, placebo-controlled trial (JADE MONO-2) – 391 patients (≥ 18 years) – 3 groups of patients: abrocitinib 100 mg ID (n = 158), abrocitinib 200 mg ID (n = 155), placebo (n = 78) |
Coprimary endpoint: Investigator Global Assessment response: score 0 (clear) or score 1 (almost clear) ≥ 75% improvement in EASI (EASI-75) score from baseline | Investigator Global Assessment response and EASI-75 at week 12: abrocitinib 100 mg ID group: 28.4% and 44.5%; abrocitinib 200 mg ID group: 38.1 and 61%; placebo group: 9.1 and 10.4%, respectively | AE was reported in 62.7% of patients in abrocitinib 100 mg ID group, 65.8% of patients in the abrocitinib 200 mg ID group, and 53.8% of patients in the placebo group Most common AE in the abrocitinib 100 and 200 mg group: nausea (n = 12, n = 22, respectively) and nasopharyngitis (n = 20, n = 12, respectively) SAE related to treatment: two in the 100 mg group (one developed pneumonia, and one developed herpangina) and two in the placebo group (one case of eczema herpeticum, and one case of staphylococcal infection). No SAE related to treatment was reported in the 200 mg group | |
| Blauvelt et al.80 | Phase 3, randomized, multicentric, responder-enriched, double-blind, placebo-controlled trial (REGIMEN) 1,233 patients (≥ 18 years) Three periods: (a) Induction period: 12-week monotherapy with abrocitinib 200 mg ID to determine response (b) Maintenance- withdrawal period: induction period responders (IGA 0/1 response and EASI-75 response) were randomly assigned in a 1:1:1 ratio to blinded abrocitinib (200 or 100 mg ID) or placebo for 40 weeks (c) Patients with a flare during the maintenance period receive rescue treatment (abrocitinib 200 mg plus topical therapy for 12 weeks) | Primary endpoint: loss of response requiring rescue medication during the maintenance period | Of 1233 patients, 798 responders to induction (64.7%) were randomly assigned The flare probability during maintenance was 18.9, 42.6, and 80.9% with abrocitinib 200 mg, abrocitinib 100 mg, and placebo, respectively Among patients with flare in the abrocitinib 200 mg, abrocitinib 100 mg, and placebo groups, 36.6, 58.8, and 81.6% regained IGA response, respectively, and 55.0, 74.5, and 91.8% regained EASI-75 response, respectively, with rescue treatment | AE reported during maintenance was 63.2 and 54.0% of patients receiving abrocitinib 200 and 100 mg, respectively | |
| Shi et al.81 | Phase 3, randomized, long extension study (JADE EXTEND) 223 patients (≥ 18 years) Patients with moderate-to-severe atopic dermatitis were randomized to receive abrocitinib 200 mg or 100 mg once daily (JADE EXTEND) after dupilumab every other week (JADE COMPARE) during 16 weeks. The final subcutaneous dose was administered at week 14 and patients received oral placebo until week 20, at which time patients were permitted to enter JADE EXTEND. | Primary endpoints: IGA, EASI and PP-NRS at baseline and at week 2, 4 and 12. | At week 12, among dupilumab responders: EASI-75 was achieved in 93.5 and 90.2% of patients who received 12 weeks of abrocitinib 200 and 100 mg, respectively PP-NRS 4-point improvement was achieved in 89,7% and 81,6%, respectively. Among patients who achieved EASI-75 but not EASI-90 with dupilumab in JADE COMPARE, 64.7% of patients treated with abrocitinib 200 mg and 54.1% of patients treated with abrocitinib 100 mg achieved EASI-90 at week 12. At week 12, among dupilumab nonresponders: EASI-75 was achieved with abrocitinib 200 mg and 100 mg in 80.0, and 67.7%. PP-NRS 4-point improvement was achieved in 77.3 and 37.8%, respectively. | – The most common treatment-emergent EA with abrocitinib 200 mg and abrocitinib 100 mg were nasopharyngitis (11.0 and 6.9%, respectively), nausea (8.2 and 0%, respectively), acne (6.8 and 2.3%, respectively) and headache (6.8 and 0.8%, respectively). SAE related to treatment: none with abrocitinib 200 mg; one patient who received abrocitinib 100 mg had eczema herpeticum. | |
| Baricitinib2 | Simpson et al.82 | – Two independent, multicentric, double-blind, phase 3, randomized, placebo-controlled trials (BREEZE-AD1 and BREEZE-AD2). – 624 patients in BREEZE-AD1 and 615 patients in BREEZE-AD2 (≥ 18 years) – 4 groups of patients: once-daily placebo, baricitinib 1 mg, 2 mg, 4 mg in monotherapy. |
Primary endpoint: – vIGA 0/1 with ≥ 2 improvement from baseline at week 16 of 4, 2 mg baricitinib and placebo |
BREEZE-AD1: vIGA 0/1 response at week 16 was achieved in 16.8, 11.4, and 4.8% patients with baricitinib 4, 2, and placebo, respectively (p < 0.001, p < 0.05) BREEZE-AD2: vIGA 0/1 response at week 16 was achieved in 13.8%, 10.6%, and 4.5% patients with baricitinib 4, 2, and placebo, respectively (p = 0.01, p < 0.05) | Tretament-emergent AEs were reported in 54, 54, 58, and 58% of patients and 56, 53, 58, and 54% of patients on placebo, 1, 2, and 4 mg in BREEZE-AD1 and BREEZE-AD2, respectively Nasopharyngitis, upper-respiratory tract infections, CPK elevations and headaches were the most frequent AEs (> 2% in any group) |
| Reich et al.83 | – Phase 3, randomized, multicentric, responder-enriched, double-blind, placebo-controlled trial (BREEZE-AD7) – 329 patients (≥ 18 years) – 3 groups of patients: once-daily placebo (n = 109), baricitinib 2-mg (n = 109), 4-mg (n = 111). – All patients were on concomitant topical corticosteroids therapy and patients were permitted to use topical calcineurin inhibitors. |
Primary endpoint: - vIGA 0/1 with ≥ 2 improvement from baseline at week 16 of 4mg, 2 mg baricitinib and placebo | vIGA 0/1 response at week 16 was achieved in 31, 24, and 15% of patients with baricitinib 4, 2, and placebo, respectively (p = 0.04 for 4 mg group and p = 0.08 for 2 mg group) | Tretament-emergent AEs were reported in 58, 56 and 38% of patients in baricitinib 4, 2mg, and placebo group, respectively. The most common AE were nasopharyngitis, upper-respiratory tract infections and folliculitis (≥ 2% in any group). SAE were reported in 4% in 4 mg group, 2% in 2 mg group and 4% in the placebo group. | |
| Silverberg et al.84 | Phase 3, multicenter, double-blind, long-term extension study (BREEZE-AD3) 1,239 patients (≥ 18 years) Responders and partial responders (vIGA score of 0, 1 or 2) at BREEZE-AD1/BREEZE-AD2 completion remained on originally assigned treatment for 52 weeks (68 total weeks of continuous therapy). | Primary endpoint: – vIGA 0/1 at weeks 16, 36, and 52. |
The proportion of baricitinib, 4 mg, responders and partial responders (n = 70) achieving or maintaining vIGA-AD (0.1) was stable: 45.7% at baseline (week 16 of continuous therapy) and 47.1% at week 68 of continuous therapy. The proportion of baricitinib, 2 mg, responders and partial responders (n = 54) achieving or maintaining vIGA-AD (0.1) was mostly stable to slightly increased (week 16, 46.3%; week 68, 59.3%). | – | |
| Upadacitinib3 | Guttman-Yassky et al.6 | – Phase 3, randomized, multicentric, double-blind, placebo-controlled trial (Measure Up 1 and Measure Up 2) – 847 patients assigned Measure Up 1 study and 836 patients assigned Measure Up 2 study (≥ 12 years) – 3 groups of patients: Measure Up 1: upadacitinib 15 mg (n = 281), upadacitinib 30 mg (n = 285) or placebo (n = 281) Measure Up 2: upadacitinib 15 mg (n = 276), upadacitinib 30 mg (n = 282) or placebo (n = 278) |
Coprimary endpoints: - Investigator Global Assessment response: score 0 (clear) or score 1 (almost clear) - ≥ 75% improvement in EASI (EASI-75) score from baseline | Measure Up 1: EASI-75 and vIGA 0/1 at week 16: upadacitinib 15 mg 70 and 48%; upadacitinib 30 mg 80% and 62%; placebo 16 and 8%, respectively (all p < 0.001). Measure Up 2: EASI-75 and vIGA 0/1 at week 16: upadacitinib 15 mg 60 and 39%; upadacitinib 30 mg 73 and 52%; placebo 13 and 5%, respectively (all p < 0.001). | Any treatment-emergent AE and SAE: Measure Up 1: 63 and 2% in upadacitinib 15 mg group; 73 and 3% in upadacitinib 30 mg: 59 and 3% in placebo, respectively. Measure up 2: 60 and 2% in upadacitinib 15 mg group; 61 and 3% in upadacitinib 30 mg; 53 and 3% in placebo, respectively. The most frequently reported treatment-emergent AE were acne, upper respiratory tract infection, nasopharyngitis, headache, elevation in creatinine phosphokinase and AD. |
| Silverberg et al.85 | – Phase 3, randomized, multicentric, double-blind, placebo-controlled trial (AD up) – 901 patients (> 12 years) – Three groups of patients: 300 were randomized to upadacitinib 15 mg + TCS, 297 to upadacitinib 30 mg + TCS and 340 to placebo + TCS. At week 16, a total of 283 placebo + TCS treated patients were rerandomized: 144 to upadacitinib 15 mg + TCS and 139 to upadacitinib 30 mg + TCS throught week 52 |
Coprimary endpoints: – Investigator Global Assessment response: score 0 (clear) or score 1 (almost clear) – ≥ 75% improvement in EASI (EASI-75) score from baseline |
EASI-75 and vIGA 0/1 at week 16: upadacitinib 15 mg + TCS 64.3 and 39.3%; upadacitinib 30 mg + TCS 76.9 and 58.4%; placebo + TCS 26.3 and 1.2%, respectively EASI-75 and vIGA 0/1 at week 52: upadacitinib 15 mg + TCS 50.8 and 33.5; upadacitinib 30 mg + TCS 69.0 and 45.2%, respectively | The most frequently reported treatment emergent AE were acne, nasopharyngitis, blood creatine phosphokinase increase, dermatitis atopic and upper respiratory tract infection Rates of SAE were similar between treatment groups (8.0 and 8.1 E/100 PY with upadacitinib 15 mg + TCS and upadacitinib 30 mg + TCS, respectively |
EASI-75: improvement in EASI (Eczema Area and Severity Index) score from baseline; ID: each day; AE: adverse effects; SAE: serious adverse effects; vIGA: validated Investigator Global Assessment response-defined as patients who achieved IGA 0 (clear)/1 (almost clear); PP-NRS: peak pruritus numerical rating scale; CPK: creatine phosphokinase; TCS: topical corticosteroids; E: events; PY: person-years,1 In both monotherapy studies (MONO-1 and MONO-2) and in the combination therapy study (COMPARE), a significantly greater proportion of patients achieved at least a PP-NRS 4-point improvement (PP-NRS4 responders were patients with ≥ 4-point improvement in PP-NRS from baseline) with 100 or 200 mg once daily abrocitinib compared with placebo. This improvement was observed as early as Week 2 and persisted through Week 12. Additionally, abrocitinib significantly improved patient-reported outcomes, including itch, sleep (SCORAD Sleep VAS), AD symptoms (POEM), quality of life (DLQI) and symptoms of anxiety and depression (HADS), at 12 weeks compared to placebo5,78,79. In REGIMEN study, the probability of maintenance of response during 40 weeks was higher for abrocitinib 200 mg versus 100 mg and for both abrocitinib doses verus placebo. These observations support continuous abrocitinib 200 mg monotherapy as the most effective option for maintaining disease control80,2. In both monotherapy studies (BREEZE-AD1 and BREEZE-AD2) and in the concomitant TCS study (BREEZE-AD7), baricitinib 4 mg significantly improved patient-reported outcomes, including itch NRS, sleep (ADSS), skin pain (skin pain NRS), quality of life (DLQI) and symptoms of anxiety and depression (HADS), at 16 weeks compared to placebo82,83,3. The week-16 results from the Measure Up 1 and Measure Up 2 studies demonstrated that upadacitinib was superior to placebo across assessments of disease activity, itch, skin pain, and impact of AD on quality of life. Clinically and statistically significant improvements in itch were observed as early as 2 days after the first dose of upadacitinib, and skin clearance (≥ 75% improvement in Eczema Area and Severity Index [EASI-75]) was observed as early as week 26.
Topical JAK inhibitors
Topical treatments, including corticosteroids and calcineurin inhibitors, are considered standard-of-care therapy for most patients with AD; however, their clinical benefit is often limited by their anatomic use restrictions and local AEs, including skin atrophy, striae, and/or application site reactions. Long-term application of these drugs, particularly in sensitive areas, is not recommended owing to safety/tolerability. Thus, the need remains for a nonsteroidal topical therapy that is highly effective, well tolerated, and provides rapid and durable resolution of inflammatory lesions and pruritus11.
JAK inhibitors have also been developed as a topical treatment option and are gaining attention as a treatment option for various skin diseases. Mean plasma concentrations below those achieved with the oral formulation, low incidence of AEs, and stable hematologic markers support a lack of systemic toxicity with topical JAK inhibition12.
Delgocitinib, a first-generation pan-JAK inhibitory profile that inhibits all the JAK activities in enzyme assays, inhibits the activation of inflammatory cells, such as T cells, B cells, monocytes, and mast cells, and improves skin barrier dysfunction13. In a 52-week, long-term, open-label study for patients aged 16 years with mild-to-severe AD, treated with delgocitinib 0.5% ointment, the proportion of patients with mEASI-50 at week 4, 24, and 52 were 31.5, 42.3, and 51.9%, respectively. While those with mEASI-75 at weeks 4, 24, and 52 were 10.9, 22.7, and 27.5%, respectively. AEs and treatment-related AEs were reported in 69.0% of the patients. The most common AE was nasopharyngitis, followed by contact dermatitis, acne, and application site folliculitis. Study discontinuations due to AEs occurred in 17 patients (3.4%), and the most common AEs leading to study discontinuation were contact dermatitis in five patients (1.0%) and application site irritation in three (0.6%)14.
In a phase 2a study on adult patients with mild-to-moderate AD using 2% tofacitinib ointment, the mean percentage change in EASI scores from the baseline was significantly greater (p < 0.001) for tofacitinib (81.7%) compared to the vehicle group (29.9%) at week 4. Meanwhile, patients treated with tofacitinib ointment showed significant improvements in pruritus by day 2. Safety/local tolerability was generally similar for both treatments, although more adverse events were observed for a vehicle compared to tofacitinib15.
Ruxolitinib (RUX) cream, a selective inhibitor of JAK1/2, demonstrated potent anti-inflammatory effects versus vehicle with rapid and sustained itch control in these phase three studies in patients with AD. Two double-blind, randomized, vehicle-controlled trials of identical design (TRuE-AD1 and TRuE-AD2) enrolled a total of 1,249 adult and pediatric subjects aged 12 and older. Patients were randomized 2:2:1 to twice-daily RUX 0.75% cream, 1.5% RUX cream, or vehicle cream for 8 continuous weeks. The primary efficacy endpoint was the proportion of subjects at week 8 achieving IGA treatment success (IGA-TS), defined as a score of 0 (clear) or 1 (almost clear) with ≥ 2-grade improvement from baseline. Efficacy was also assessed using a ≥ 4-point improvement in Itch NRS. In TRuE-AD1/ TRuE-AD2 significantly more patients achieved IGA-TS with 0.75% RUX cream (50.0/39.0%) and 1.5% RUX cream (53.8/51.3%) vs vehicle (15.1/7.6%; p < 0.0001) at week 8. Significantly more patients in TRuE-AD1 and TRuE-AD2 achieved EASI-75 at week 8 with 0.75% RUX (56.0 and 51.5%, respectively) and 1.5% RUX (62.1 and 61.8%) vs vehicle (24.6 and 14.4%). Application site reactions were infrequent (< 1%) and lower with RUX versus vehicle. The most common treatment-related AE was application site burning sensation, which was observed primarily with vehicles (4.4%; 0.75% RUX-0.6%; 1.5% RUX-0.8%). Additionally, significant itch reductions vs vehicle were reported within 12 h of the first application of 1.5% RUX, and further reductions were observed over 8 weeks11. Therefore, it is thought that topical JAK inhibitors rapidly improve AD rash and symptoms relatively safely.
Currently, OPZELURA® (1.5% RUX cream) is approved in the United States (US) but not in Europe for the treatment of AD, which configurated a major milestone for the treatment of these patients. Table 1.
Alopecia areata
Alopecia areata (AA) is a chronic immune-mediated disease characterized by nonscaring hair loss with clinical heterogeneity16. The exact etiology of this disease is not yet elucidated, and it is thought that the hair follicle bulb immune privilege collapse is critical in the pathophysiology of the disease. Although it is not yet fully understood what causes immune privilege to collapse, triggering factors suppress immune privilege guardian expression and locally activate the innate immune system mostly via CD8 + NKG2D T cells, leading to subsequent interferon-γ (IFN-γ) production and major histocompatibility complex class I upregulation that further contribute to immune privilege breakdown16.
Current treatment options for AA, including topical, systemic, and injectable interventions, aim to immunosuppress or immunomodulate the activity of the disease, with a generally unsatisfying response and high relapse rate. Additionally, the available therapeutic options do not seem to influence the long-term course of the disease and thus, an urgent need remains for novel, more effective therapies16. The nonspecific nature of these treatment modalities, along with their variable efficacy demands effort to achieve more targeted therapies that can better target the pathways involved in the disease process17.
Local inflammation in AA is largely mediated by the JAK-STAT pathway; in this disease, there is an overexpression of proinflammatory cytokines that signal through their receptors via JAK-STAT. Additionally, this intracellular signaling network has an important role in mediating and maintaining the cytotoxic CD8 + NKG2D + T-cell reaction, which represents a driving component for AA pathogenesis. In alopecia areata, JAK1/2 and JAK1/3 lead to T-cell mediated inflammatory responses, which promote IFN-γ and IL-15 production in hair follicles and further amplify the inflammatory feedback loop16,17. Thus, it is not surprising that JAK inhibitors represent an emerging treatment option for AA16,17.
There have been a number of case reports and small clinical trials reporting promising outcomes of JAK inhibitors tofacitinib, RUX, and baracitinib for alopecia areata18. The efficacy and safety of baricitinib were assessed in two randomized, double-blind, placebo- controlled trials (BRAVE-AA1 and BRAVE-AA2) that enrolled a total of 1200 patients with AA and led to its recent approval (2022) by the EMA and the Food and Drug Administration (FDA) (Table 1), which constituted a major milestone for the treatment of AA (Table 1)9,19,20.
Systemic Janus Kinase inhibitors
There have been several publications of clinical research studies on the efficacy of tofacitinib (oral or topical preparation) in the treatment of AA and its variants. Liu et al. 2017, conducted a retrospective study consisting of 90 patients (aged 18-70 years) diagnosed with AA, alopecia universalis, or alopecia totalis. Patients were administered oral tofacitinib 5 or 10 mg twice daily (bid) with or without prednisolone and were evaluated using the severity of alopecia tool (SALT) scoring system at baseline and then at various treatment intervals for 4-8 months. As such, 58% of patients achieved greater than 50% change in SALT score over 4-18 months of treatment, 20% of patients were complete responders and had full regrowth with > 90% reduction in SALT, whereas 56.9% were intermediate to moderate responders (25 patients intermediate and 12 patients moderate) with 51-90% reduction in SALT for intermediate responders and 6-50% reduction in SALT for moderate responders. Additionally, 23.2% of patients were identified as nonresponders with ≤ 5% reduction in SALT21.
Multiple case reports have examined the use of RUX in the treatment of AA. In an open-label clinical trial, Mackay-Wiggan et al. aimed to investigate RUX 20 mg orally bid in the treatment of moderate-to-severe AA. All patients received RUX for 3-6 months, followed by a 3-month observational phase to assess treatment response durability. About 9 out of 12 (75%) had significant hair regrowth and achieved the primary outcome of at least 50% regrowth, with seven of the nine responders achieving over 95% regrowth by the end of treatment22. The durability of responses was assessed in the 3-month follow-up period of treatment. Around three out of nine responders noted shedding beginning at week 3 following RUX discontinuation and had marked hair loss at week 12 off the drug; however, hair loss did not reach baseline levels. Around six out of nine responders reported increased shedding without major hair loss22.
Baricitinib is now approved for the treatment of AA (Table 1)9. The efficacy and safety of baricitinib once daily were assessed in two randomized, double-blind, placebo-controlled trials (BRAVE-AA1 and BRAVE-AA2) that enrolled a total of 1,200 patients with AA who had at least 50% scalp hair loss as measured by the SALT for > 6 months. In both phase III 36-week studies with extension phases up to 200 weeks, patients were randomized to placebo, 2 or 4 mg baricitinib in a 2:2:3 ratio. Both trials assessed the proportion of patients who achieved at least 80% scalp hair coverage (SALT score of ≤ 20) at week 36 as the primary endpoint. Other outcomes at week 36 included the proportion of patients who achieved at least 90% scalp hair coverage (SALT score of ≤ 10), patients with Scalp Hair Assessment PRO score of 0 or 1 with at least a 2-point reduction on the 5-point scale, and assessments of eyebrow and eyelash hair loss. The estimated percentage of patients with a SALT score of 20 or less at week 36 was 38.8% with 4 mg baricitinib, 22.8% with 2 mg baricitinib, and 6.2% with placebo in BRAVE-AA1 and 35.9, 19.4, and 3.3%, respectively, in BRAVE-AA2. In BRAVE-AA1, the difference between 4 mg baricitinib and placebo was 32.6% points, and the difference between 2 mg baricitinib and placebo was 16.6% points (p < 0.001 for each dose vs placebo). In BRAVE-AA2, the corresponding values were 32.6% and 16.1% points (p < 0.001 for each dose vs placebo). Most patients in whom the primary outcome was met had SALT scores of 10 or less at week 36. Secondary outcomes for baricitinib at a dose of 4 mg but not at a dose of 2 mg generally favored baricitinib over placebo20. In the study BRAVE-AA2, patients who had received baricitinib 4 mg once daily since the initial randomization and achieved a SALT of ≤ 20 at week 52 were rerandomized in a double-blind manner to continue 4 mg once daily or reduced dose to 2 mg once daily. The results show that 96% of the patients who remained on baricitinib 4 mg and 74% of the patients who were rerandomized to baricitinib 2 mg maintained their response at week 769.
Topical JAK inhibitors
Very limited studies have addressed the efficacy of topical JAK inhibitors in the treatment of AA17. Liu et al. in 2018 conducted an open-label, single-centre pilot study of 10 patients with AA to assess the efficacy and safety of topical tofacitinib. Patients were treated with tofacitinib 2% ointment applied bid for 6 months and were evaluated periodically for regrowth using SALT. Regrowth occurred in three patients at week 24, one patient had a SALT score percent change of 61%, while the other two patients had 18% and 25% change and seven patients had no regrowth23. The topical efficacy of RUX has not been well studied; there are only a few data available and the results shown are still not very encouraging24-26.
Treatment results by route of administration and sustainability of response with JAK inhibitors
Phan et al. conducted a meta-analysis that sought to determine the expected response of AA to JAK inhibitor therapy and factors that influence response and recurrence rates. From 30 studies and 289 cases, there were 72.4% responders, 45.7% good responders, and 21.4% partial responders. The mean time to initial hair growth was 2.2 ± 6.7 months. The oral route, regardless of the specific agent of oral JAK inhibitor used in treatment, was significantly associated with four times higher odds of achieving a response when compared to topical therapy18. Oral JAK inhibitor treatment was also associated with seven times higher odds of achieving a good response (50-100% regrowth) than a partial response (5-50% regrowth) compared to topical treatment, with no difference between tofacitinib, RUX, or baracitinib18. Thus, so far, oral JAK inhibitors have demonstrated a higher efficacy in the treatment of AA than topical JAK inhibition18.
The sustainability of treatment results with JAK inhibitors used for AA management has been a topic of concern for many researchers and clinicians17. In clinical practice, it has been observed that AA frequently recurs after cessation of JAK inhibitors therapy17,18,27. In a systemic review and meta-analysis, it has been observed that all cases of relapse were associated with cessation of therapy, on average, after 2.7 months. These results suggest that the therapeutic response may only be maintained whilst the patient is on JAK inhibitor treatment and that once ceased, relapse of AA may be expected within 3 months18.
Vitiligo
Vitiligo is an acquired, idiopathic, and multifactorial autoimmune disorder characterized by patchy depigmentation in the skin, hair or both28. Depigmentation that characterizes vitiligo is caused by progressive melanocyte destruction. Activated CXCR3+ CD8+ T cells promote melanocyte detachment and apoptosis through INF-γ (interferon-gamma), which stimulates the JAK/STAT-1 pathway leading to the expression of INF-stimulated genes with further recruitment of CXCR3+ CD8+ T cells and the formation of a positive-feedback loop. The IFN-γ-chemokine axis has been identified as a potential pathway in the initiation and progression of vitiligo28-30. JAK inhibitors are a promising targeted treatment for vitiligo.
Systemic and topical JAK inhibitors
In vitiligo mouse models, neutralization of IFN-γ with antibody prevents CD8+ T-cell accumulation and depigmentation, suggesting a therapeutic potential for this approach29. The JAK/STAT pathway plays a central role in vitiligo and inhibition of this intracellular signaling network has been shown to block IFN-γ signaling and contribute to repigmentation in individuals with vitiligo31.
Tofacitinib can induce significant repigmentation in patients with vitiligo; however, concomitant stimulation of melanocytes via skin exposure to ultraviolet light appears to be required to achieve repigmentation32. Baricitinib differs from tofacitinib in that it preferentially inhibits JAK1/2 rather than JAK 3. Given IFN-γ is mediated via JAK1/2, it has been suggested that JAK inhibitors, which predominantly inhibit these JAKs may be more effective than others32. A small number of studies have addressed the efficacy of tofacitinib and baricitinib in the treatment of vitiligo (Table 4) and further research is necessary to determine their safety and efficacy.
Oral RUX interferes with IFN-γ signaling by preferential inhibition of JAK 1 and JAK 2 and has been shown to improve skin conditions in some studies28,31. Rapid skin repigmentation on oral RUX was observed in a patient with coexistent vitiligo and AA, with marked declines in serum CXCL10 levels (chemokine ligand 10) after administration31. Chemokine ligand 10 (CXCL10), an IFN-γ-induced chemokine, was shown to be critical for autoreactive T-cell recruitment to the skin during the progression and maintenance of vitiligo33,34. However, topical administration resulted in a higher concentration in the epidermis and dermis, resulting in near-complete inhibition JAK/STAT signaling in this tissue35. In contrast, only partial inhibition of downstream signaling was predicted to occur after oral dosing. Therefore, dermis concentrations of RUX cream are fully effective, whereas corresponding plasma concentrations are negligible. Consequently, this distribution profile should maximize the efficacy of RUX cream in the skin while minimizing the potential for deleterious systemic effects35. Interestingly, according to meta-analyses, it also seems that topical JAK inhibitor formulations perform comparably to oral counterparts36. Two double-blind, randomized, vehicle-controlled trials (TRuE-V1 and TRuE-V2) aimed to evaluate the efficacy and safety of RUX cream for the treatment of vitiligo (Table 4). In both trials, subjects were randomized 2:1 to treatment with 1.5% RUX cream bid or vehicle cream bid for 24 weeks, followed by an additional 28 weeks of treatment with 1.5% RUX cream bid for all subjects37,38. Lesions on the face were assessed with the facial vitiligo area scoring index (F-VASI) and lesions on the total body (including the face) were assessed with the total body vitiligo area scoring index (T-VASI). At week 52, approximately 50% of patients achieved ≥ 75% improvement in the F-VASI75 compared to the F-VASI75 improvement from baseline reported for these patients at week 24 (the primary endpoint of the study), which was approximately 30%. Additionally, a greater proportion of patients at week 52 achieved ≥ 50% improvement in T-VASI50, and over one-third of patients achieved a vitiligo noticeability scale response. Further improvement in percentage change from baseline in facial body surface area with the application of RUX cream was also observed. RUX cream was well tolerated, the most common treatment-related TEAEs were application site acne and pruritus, no serious TEAEs were considered related to treatment and there were no significant changes in hemoglobin or platelet levels37,38. Based on the exciting results of TRuE-V1 and TRuE-V2, RUX was recently approved by the FDA for the topical treatment of ≥ 12 years of patients with nonsegmental vitiligo (Table 1)39.
Table 4 Summary of reports with tofacitinib, baricitinib, and ruxolitinib in vitiligo
| Authors | Treatment | Patient | Results | Safety | |
|---|---|---|---|---|---|
| Tofacitinib | Brittany et al.86 | Oral tofacitinib 5 mg every other day for 1 week, then 5 mg daily | 1 patient (white macules and patches involving the forehead, trunk, and extremities involving 10% of body surface area) | After 5 months, repigmentation of the forehead and hands was nearly complete, and the remaining involved areas demonstrated partial repigmentation. Approximately 5% of the total body surface area remained depigmented | No adverse effects |
| Liu et al.32 | Oral tofacitinib, 5-10 mg daily or twice daily for an average of 9.9 months | 10 patients (eight patients had generalized vitiligo and two patients had primarily acral involvement, with BSA 1-10%) | A mean decrease of 5.4% BSA involvement with vitiligo was observed in five of 10 patients, at sites of either sunlight exposure or low-dose narrowband ultraviolet B phototherapy | The most common adverse event was upper respiratory infection in two patients. There were no serious adverse events. | |
| McKesey et al.87 | 2% tofacitinib cream twice daily in conjunction with narrowband ultraviolet B (NB-UVB) therapy twice weekly over a period of 3 ± 1 months | 11 patients (the mean facial VASI was 0.80 at baseline) | The mean facial VASI was 0.23 at follow up, which is a mean improvement of 70% (range 50-87%). Mean time to follow-up was 112 days | – | |
| Mobasher et al.40 | Tofacitinib 2% cream twice daily to target areas | 16 patients (three patients had focal facial vitiligo, two had focal nonfacial vitiligo, while 11 had generalized vitiligo) | 13 experienced repigmentation with 4 patients experiencing > 90% repigmentation; 5 patients experiencing 25-75% repigmentation; and 4 patients experiencing 5-15% repigmentation. Two patients experienced no change. Facial lesions improved more than non-facial lesions | There were no serious adverse events. | |
| Baricitinib | Mumford et al.88 | Oral baricitinib 4 mg daily | 1 patient | At follow-up 8 months after the commencement of baricitinib, almost complete repigmentation of the hands and forearms was observed | No adverse events |
| Harris et al.31 | Oral RUX 20 mg BID | 1 patient with widespread, near-complete depigmentation of his face, along with lesions on his trunk and extremities. He also had patches of nonscarring alopecia on his scalp and extremities. | At week 12: 85% scalp hair compared with 63% at baseline At week 20: improvement of facial pigmentation from 0.8% to 51% 12 weeks after discontinuing RUX, while his hair regrowth was maintained, much of the regained pigment had regressed | – | |
| Ruxolitinib | Rothstein et al.41 | Topical RUX 1.5% cream BID | 12 Patients with a minimum of 1% affected body surface area of vitiligo | At week 20: VASI improved 23%. Significant repigmentation (76%) in facial vitiligo in 4 patients | There were no severe or lasting side effects |
| Rosmarin et al.37,38 | 1,5% RUX cream twice daily or vehicle cream twice daily (BID) for 24 weeks followed by an additional 28 weeks of treatment with 1.5% RUX cream BID for all subjects | Two double-blind, randomized, vehicle-controlled trials of identical design (TRuE-V1 and TRuE-V2) Enrolled a total of 674 adult and pediatric subjects aged 12 years and older | Primary efficacy endpoint: the proportion of subjects achieving at least 75% improvement in F-VASI (F-VASI75) at Week 24: 29.9/29.9% in RUX cream group and 7.5/12.9% in vehicle group, in TRuE-V1 and TRuE-V2, respectively Other endpoints: at week 52, approximately 50% of patients achieved F-VASI75 | Treatment emergent adverse effects: 37.6/33.0% in vehicle group and 45.7/49.6% in TRuE-V1 and TRuE-V2, respectively |
BID: twice daily; BSA: body surface area; F-VASI75: ≥ 75% improvement in the facial vitiligo area scoring index; VASI: vitiligo area severity index.
It appears that vitiligo located on the face responds more robustly to therapy than nonfacial lesions36,40,41. Rothstein et al. conducted an open-label trial of bid RUX 1.5% cream in 12 patients and showed a 23% improvement in VASI scores at week 20. Four patients had significant facial involvement at baseline and had a 76% improvement in facial VASI scores41.
Several studies have shown superior repigmentation rates in patients with JAK inhibitors and concomitant UV exposure. Joshipura et al. reported significant repigmentation in sun-exposed areas in two patients treated with either topical RUX 1.5% bid or oral tofacitinib 5 mg bid42. Another retrospective case series of 10 patients treated with oral tofacitinib showed that five patients achieved some repigmentation at sites of sun exposure or UVB phototherapy32. Phan et al. found that a good response rate or repigmentation rate > 50% was found in 57.8% of patients treated with JAK inhibitors, but when used concurrently with phototherapy, the good response rate improved to 88.9%36. These data suggest that substantial repigmentation in vitiligo using JAK inhibitor may require photoactivation to stimulate melanocytes, which supports a multimodal therapeutic approach.
A few registered ongoing trials are focusing on the use of second-generation JAK inhibitors in the treatment of patients with vitiligo (https://clinicaltrials.gov/).
JAK inhibitors appear to be a promising treatment for vitiligo and the recent approval of RUX in the US was an exciting milestone for the treatment of these patients; however, further studies are required to confirm efficacy, establish safety, and investigate the durability of repigmentation.
Psoriasis vulgaris
Psoriasis (PSO) is a common chronic inflammatory skin disease with well-defined pathogenesis in which IL-23/Th17 signaling axis plays a central role. In the last years, numerous targeted treatments have been developed for PSO2. The majority of those affected with PSO have mild-to-moderate forms and are usually treated with topical therapy, whereas phototherapy and systemic therapies are used for those with severe disease43. The implication of multiple cytokines like IL-6, IL-22, IL-23, or INF-γ in PSO pathogenesis that signal through the JAK/STAT pathway suggests that the inhibition of JAKs could be a viable therapeutic option for this disease2,43. Additionally, recent studies have shown that PSO is mainly a JAK3 and JAK1-driven disease with a predominance of STAT3 signaling44. STAT3 mediates the signal of most cytokines that are involved in disease pathogenesis, including the central IL-23/IL-17/IL-22 axis, and active STAT3 is found in psoriatic skin45,46. Despite the recent availability of effective biological agents (monoclonal antibodies) against IL-17 and IL-23, which have radically changed the current standard of disease management, the possibility of targeting either STAT3 itself or the family of upstream activators JAKs offers additional therapeutic options46. Additionally, JAK inhibitors are less expensive when compared to biologics3.
Systemic JAK inhibitors
Tofacitinib, a pan-JAK inhibitor with a predominant anti-JAK3 effect, is the most studied oral JAK inhibitor for the treatment of chronic plaque PSO3. Two-phase three randomized place-controlled trials (OPT Pivotal 1 and OPT Pivotal 2) demonstrated the efficacy of tofacitinib 5 or 10 mg bid over placebo in patients with moderate to severe PSO, improvement in nail PSO was also observed47. Nevertheless, tofacitinib 10 mg bid was more effective compared to 5 mg bid dosage48. Bissonnette et al. showed that continued tofacitinib worked best in PSO and that treatment discontinuation was associated with a risk of relapse; however, among relapsers, up to 60% recaptured response with tofacitinib49. Tofacitinib is approved by the FDA and EMA for the treatment of psoriatic arthritis, but the approval was not extended for PSO (Table 1). It is difficult to see tofacitinib achieving approval for treating PSO at the 5 mg bid dose, and there are safety concerns, as noted by the FDA, of the more effective 10 mg bid dose48. Numerous oral JAK inhibitors, such as abrocitinib, solcitinib, itacitinib, peficitinib, were tested for the treatment of PSO50-53.
Tyrosine kinase (TYK2) inhibitors, such as deucravacitinib, are promising therapeutic options for the treatment of PSO, given that TYK2 is responsible for mediating immune signaling of IL-12, IL-23, and type I interferons without interfering with other critical systemic functions as other JAK proteins do54-57. Unlike TYK2, JAK1, 2, and 3 are responsible for mediating a series of signals that support broader systemic responses, such as hematopoiesis, myelopoiesis, lipid metabolism, and bone regulation. Consequently, JAK1, 2, and 3 inhibitors, such as tofacitinib, baricitinib, and ruxolitinib, a raised safety concerns and their clinical research in PSO have been mostly abandoned due to their unfavorable efficacy/safety ratio58.
POETYK PSO-1 and 2 enrolled 1686 patients with moderate-to-severe PSO. After 16 weeks, in both studies, over 50% of patients treated with deucravacitinib reached PASI75, which was significantly superior to placebo and apremilast. These results were maintained through week 52, with over 65% of patients achieving PASI75 at this point in POETYK PSO-157,59. A reduction in signs and symptoms was also reported by patients, with a greater impact on itch. It was well tolerated and safe57,59.
Deucravacitinib was recently approved by the US for the treatment of patients with moderate to severe plaque PSO and had the potential to become an efficacious, safe, and well-tolerated treatment (Table 1)60. Being an oral drug and an IL-23 inhibitor, its approval may have a great impact on clinical practice. Nevertheless, further studies are needed to evaluate long-term treatment effects.
Topical JAK inhibitors
In the past decades, the major advances in PSO therapy have been in systemic agents, such as immunomodulatory and biological molecules, while topical therapies have remained relatively unchanged43. It has been recently reported that psoriatic keratinocytes overexpress JAK1 and 3, making them ideal targets for topical treatment with specific JAK inhibitors44. However, so far, the efficacy of topical JAK inhibitors for PSO is not robust2,3.
Tofacitinib showed interesting results as an oral agent for the treatment of PSO and topical therapy is being studied as well (Table 5). Although systemic concentrations of tofacitinib were found in patients treated with topical formulations, serologic levels were 40-fold lower than exposures from the lowest dose tested (2 mg bid) in a previous study of oral tofacitinib in patients with moderate-to-severe PSO61.
Table 5 Summary of reports with topical tofacitinib and ruxolitinib in psoriasis
| Authors | Clinical trial | Assessment methods | Results | Safety | |
|---|---|---|---|---|---|
| Tofacitinib | Ports et al.61 | Phase 2a study randomized, multicentric, placebo-controlled trial Adult patients with mild to moderate psoriasis (n = 71) four groups of patients: 2% tofacitinib ointment 1, vehicle 1, 2% tofacitinib ointment 2 and vehicle 2 for 4 weeks administered twice daily to a single fixed 300 cm2 treatment area containing a target plaque | Primary endpoint: percentage of change from baseline in the Target Plaque Severity Score at week 4 | At week 4: Statistically significant improvement in the target plaque severity score (TPSS) for tofacitinib ointment 2% (54.4%) vs vehicle 1 (41.5%) but not for tofacitinib ointment 2% (24.2%) vs vehicle 2 (17.2%) | No serious adverse effects (AE) reported. The most common AE: nasopharyngitis and urinary tract infections |
| Papp et al.89 | Phase 2b study randomized, multicentric, vehicle -controlled trial Adult patients with mild to moderate plaque psoriasis (n = 435) groups of patients: 1% tofacitinib ointment; 2% tofacitinib ointment; vehicle applied once or twice daily | Primary endpoint: proportion of patients with Calculated Physician’s Global Assessment (PGA-C) clear or almost clear and ≥ 2 grade improvement from baseline at week 8 and 12 | At week 8 only significantly more patients receiving 2% tofacitinib ID and 2% tofacitinib BID achieved a PGA-C response of clear or almost clear and ≥ 2 grade improvement from baseline compared with the corresponding vehicle. Response rate was 18.6 and 8.1 for 2% tofacitinib ID and vehicle QD, respectively, and 22.5 and 11.3 for 2% tofacitinib BID and vehicle BID, respectively. At Week 12, no statistically significant differences versus vehicle were seen for 2 or 1% tofacitinib by either dosing regimen | Overall, 44.2% of patients experienced treatment-emergent AEs, 8.1% experienced application site AEs, and 2.3% experienced serious AEs. The highest incidence of AEs (including application site AEs) was in the vehicle QD group. The most frequently reported AEs were nasopharyngitis, upper respiratory tract infection and PSO | |
| Ruxolitinib | Punwani et al.90 | – Phase 2, double blind, and vehicle or active comparator controlled. – Adult patients with with limited (< 20% body surface area), stable but active plaque psoriasis at the baseline (n = 28) – Patients were dosed with vehicle, 0.5 or 1.0% ruxolitinib cream once a day or 1.5% twice a day for 28 days. Additional groups included two active comparators (calcipotriene 0.005% cream or betamethasone dipropionate 0.05% cream) |
Improvements in lesion scores after 28 days of treatment | Although the 0.5% cream applied once a day appeared similar to the vehicle control in the response seen, the 1.0% cream applied once a day and the 1.5% cream applied twice a day both showed improvements in lesion scores greater than seen with the vehicle control. Mean total lesion score (scaling + redness + thickness) decreased by 53% after 28 days of application with 1.0% ruxolitinib cream compared with a 32% decrease in the vehicle-treated lesions (p = 0.033), whereas for 1.5% the mean lesion score decreased 54% compared with 32% for vehicle (p = 0.056) | No serious adverse effects. Local adverse effects: 20 with ruxolitinib; 28 with the vehicle; 40 with calcipotriene; 33% with betamethasone |
| Callis et al.91 | Phase 2b vehicle-controlled study Patients with mild-to-moderate psoriasis (n = 200) Three treatment doses: 0.5, 1, and 1.5% ruxolitinib against vehicle applied daily for 12 weeks. | Reduction in PASI scores at week 12 | At week 12: reduction in PASI scores was seen with different concentrations of ruxolitinib (37 with 0.5, 40 with 1, and 35 with 1.5%) compared to 20% with vehicle | No serious adverse effects. |
BID: twice daily; ID: each day; PGA-C: calculated Physician’s Global Assessment.
Ruxolitinib (RUX) has been tested in topical formulations to treat mild to moderate PSO with favorable results Table 5 2,3. Punwani et al. showed that transcriptional markers of immune cell lineage/activation in lesional skin were reduced by topical RUX, with correlations observed between clinical improvement and decreases in markers of T helper 17 lymphocyte activation, dendritic-cell activation and epidermal hyperplasia. Additionally, there was no significant inhibition of STAT3 in peripheral blood cells, suggesting limited systemic exposure62. In conclusion of this study, topical RUX in patients with active psoriatic lesions modulates proinflammatory cytokines62. Larger studies are needed to clearly establish the efficacy and safety profiles of topical RUX for the treatment of PSO, however, the data available suggest that it may be a promising agent.
Adverse effects and safety profile
JAK inhibitors that are currently approved for the autoimmune disease have an associated black warning box for the potential increased incidence of serious infections, mortality, malignancy, major adverse cardiovascular events (MACE), and thrombosis. This warning was added recently based on results from the ORAL Surveillance study of tofacitinib vs tumor necrosis factor α inhibitors (TNF inhibitors) in rheumatoid arthritis63. In this randomized, open-label, noninferiority, safety endpoint trial involving patients with active rheumatoid arthritis who were 50 years of age or older and had at least one additional cardiovascular risk factor, patients were randomly assigned in a 1:1:1 ratio to receive tofacitinib at a dose of 5 or 10 mg bid or a TNF inhibitor. During a median follow-up of 4.0 years, the incidences of MACE and cancer, excluding nonmelanoma skin cancer, were higher with the combined tofacitinib doses (3.4 and 4.2%, respectively) than with a TNF inhibitor (2.5 and 2.9%), the noninferiority of tofacitinib was not shown.
Safety data for tofacitinib is largely derived from clinical trials in rheumatoid arthritis and PSO, and data form RUX is from clinical trials in myelofibrosis and polycythemia vera64.
Cohen et al., showed that the risk of infection and overall mortality in patients treated with tofacitinib is not significantly different from that observed with other biologic agents65. On the contrary, the ORAL Surveillance studies showed the incidence of adjudicated opportunistic infections was higher with tofacitinib than with a TNF inhibitor, however, primarily owing to the incidence of herpes zoster and all herpes zoster (nonserious and serious)63. JAK inhibitors are associated with an increased risk of varicella-zoster virus reactivation63,66. The higher rates of herpes zoster infection among patients treated with tofacitinib may be related to its mechanism of action of tofacitinib, which involves a decrease in lymphocyte activation and proliferation. Additionally, the human antiviral defense is also associated with intact responses to type I IFN and type II IFN, which receptors signal via JAK-1. Because tofacitinib inhibits signaling through JAK-1, it is possible that such a mechanism is related to an increased risk of herpes zoster infection65. Dose-related increases in lipid levels, such as total cholesterol, LDL cholesterol, triglycerides and HDL cholesterol, were observed in clinical trials; elevations were observed at 12 weeks and are generally mild67,68.
Thrombosis, including deep vein thrombosis (DVT), pulmonary embolism (PE) and arterial thrombosis, has been reported in patients receiving JAK inhibitors used to treat inflammatory conditions. In the ORAL Surveillance study, higher rates of overall thrombosis, DVT, and PE were observed compared to those treated with TNF blockers63.
Cytopenia is another potential adverse effect of JAK inhibitors, primarily JAK2 inhibition because signaling via JAK2 is utilized by erythropoietin, thrombopoietin, and G-CSF64. In the treatment of bone marrow disorders with RUX, such as myelofibrosis and thrombocytopenia can be limiting69; however in a study of 12 patients with AA treated with RUX for up to 6 months, neither this nor other cytopenia was observed22. It may be theorized that patients with healthy bone marrows are less vulnerable to cytopenia observed with JAK2 inhibition64.
In addition, respiratory infections and gastrointestinal side effects, such as nausea and diarrhea were observed70,71. Increased levels of transaminases, creatinine phosphokinase, and serum creatinine have also been observed; these changes have generally been graded as mild71. For topical applications, acne, and pruritus are often described72.
However, the long-term safety of JAK inhibitors is still not completely understood, and as investigations of this promising drug class continue, the safety profile should become clearer. In recent years, efforts have been made to develop selective JAK inhibitors with directed targets and, consequently fewer side effects.
Possible future applications
Since the JAK/STAT signaling pathway plays a crucial role in many cytokines, a variety of inflammatory dermatological disorders may benefit from this class of immunomodulators. Currently, multiple inhibitors of the JAK/STAT pathway are being investigated for the treatment of other treatment-refractory dermatologic conditions in which activation of the JAK/STAT pathway plays a role, such as dermatomyositis, graft vs host-disease, hidradenitis suppurativa, lichen planus, lupus erythematosus, pyoderma gangrenosum, cutaneous sarcoidosis, granuloma annulare, blistering skin diseases, etc.2,64. The management of chronic hand eczema (CHE) remains a challenge; in a recent phase 2a trial, topical use of delgocitinib ointment resulted in clearance of CHE after 8 weeks of treatment in a significantly greater number of patients than vehicle and was well tolerated73. The results from this study suggest that topical delgocitinib may provide therapeutic benefits to patients with CHE with inadequate responses to topical corticosteroids73.
Conclusion
The well-established efficacy of JAK inhibitors in inflammatory disorders, particularly rheumatoid arthritis, psoriatic arthritis, and ulcerative colitis, suggests the potential of their positive effects in a myriad of dermatological dermatoses as well. Dysregulation of the JAK/STAT pathway plays a role in the pathogenesis of many dermatologic diseases, including vitiligo, alopecia areata, PSO, and AD. JAK inhibitors can either be taken orally or have also been developed as a topical treatment option which constitutes a great advantage of this drug class. In the future, JAK inhibitors could prove to be a real alternative therapy for some inflammatory skin diseases. More studies are necessary to determine the doses that will optimize the efficacy, cost-effectiveness, and safety of this drug family for potential use in skin conditions in the long-term Table 6.
Table 6 Key findings on JAK inhibitors in atopic dermatitis, alopecia areata, vitiligo and psoriasis vulgaris
| Atopic dermatitis |
|---|
| Various cytokines relevant to the pathophysiology of AD activate JAK1 containing heterodimeric receptors, thereby mediating Th2 cell differentiation. Therefore, inhibition of the JAK-STAT pathway is a desirable target to modulate a broad range of cytokines involved in the pathophysiology of AD. Several oral JAK inhibitors, including abrocitinib, baricitinib, and upadacitinib, have been shown to improve the severity and symptoms of AD. In particular, an improvement in pruritus scores was detected in the early stages after the administration of these drugs. Currently, 1.5% ruxolitinib cream is approved in the United States but not in Europe for the treatment of AD, which configurated a major milestone for the treatment of these patients. |
| Alopecia areata |
| Local inflammation in AA is largely mediated by the JAK-STAT pathway; thus, it is not surprising that JAK inhibitors represent an emerging treatment option for AA. There have been a number of studies reporting promising outcomes of JAK inhibitors; the efficacy and safety of oral baricitinib led to its recent approval by EMA, which constituted an important step in the treatment of AA. To the data, oral JAK inhibitor demonstrated a higher efficacy in the treatment of AA than topical JAK inhibitor. AA frequently recurs after cessation of JAK inhibitor therapy. |
| Vitiligo |
| IFN-γ induced expression of C-X-C-motif chemokine 10 (CXCL10) in keratinocytes has been proposed as an intermediary of vitiligo depigmentation and the IFN-γ signal transduction occurs through JAK. Thus, it was postulated that JAK inhibitors may be an important therapeutic option for vitiligo by downregulating IFN-γ-chemokine axis. Topical JAK inhibitor offers a viable therapeutic alternative to topical corticosteroids and topical calcineurin inhibitors; the beneficial effects of TJK inhibitor are most pronounced on facial skin and when combined with narrowband ultraviolet B therapy. RUX 1.5% cream was recently approved by the US for the treatment of nonsegmental vitiligo. |
| Psoriasis vulgaris |
| In PSO, the involvement of JAKs has been shown and enabled the assessment of oral and topical JAK inhibitors as therapeutics. JAK1, 2, and 3 inhibitors raised safety concerns, and their clinical research in PSO has been mostly abandoned due to their unfavorable efficacy/safety ratio. Deucravacitinib is a new oral small molecule that selectively inhibits TYK2 with promising results for PSO. Although some studies have shown encouraging results with topical JAK inhibitor, their efficacy for PSO is not robust so far. |

