Disfunção do Complexo Glicose-6-fosfátase
João Santos-Antunes, Rui Fontes
Serviço de Bioquímica, Faculdade de Medicina da Universidade do Porto
A Glicogenose tipo I é uma doença autossómica recessiva caracterizada por uma deficiência do complexo da glicose-6-fosfátase, um sistema enzimático do retículo endoplasmático responsável pela hidrólise da glicose-6-fosfato e a consequente formação de glicose nos órgãos que o possuem (fígado, rim e intestino). Esta patologia compreende essencialmente dois subtipos: a Glicogenose tipo Ia, caracterizada por um defeito na unidade catalítica do sistema e a Glicogenose tipo Ib, onde se verifica uma incapacidade do transporte da glicose-6-fosfato para o lúmen do retículo endoplasmático, onde a hidrólise ocorre. As principais alterações da Glicogenose tipo I aparecem normalmente no primeiro ano de vida e consistem num abdómen globoso (devido à hepatomegalia), hipoglicemia, hiperlipidemia, hiperuricemia e hiperlactacidemia, além da neutropenia, disfunção neutrofílica, infecções recorrentes e enterite, estas verificadas no subtipo Ib. A longo prazo podem surgir adenomas hepáticos e hepatocarcinoma, insuficiência renal, gota, osteoporose e disfunção plaquetária. Para fins diagnósticos utiliza-se a análise de mutações, combinada com as alterações clínicas e bioquímicas; para o diagnóstico pré-natal faz-se o estudo do DNA extraído de amniócitos. O tratamento actual visa, essencialmente, evitar as alterações metabólicas assim como atenuar o atraso de crescimento e a deterioração precoce da função renal; consiste, fundamentalmente, no controlo glicémico e pode ser complementado com tratamento farmacológico. Em casos de insucesso terapêutico, poderá ser ponderado o transplante hepático ou renal.
Palavras-chave: glicogenose tipo I; glicose-6-fosfátase; hipoglicemia.
Type I Glycogenosis
]]> Type I Glycogenosis is an autossomic recessive disease characterized by a dysfunction of the glucose-6-phospha-tase complex, an endoplasmic reticulum system that is responsible for the glucose-6-phosphate hydrolysis and consequent formation of glucose in liver, kidney, and intestine. This disease has essentially two subtypes: Type Ia Glycogenosis, in which a defect in the system’s catalytic unit is present, and Type Ib Glycogenosis, which is caused by a failure of the glucose-6-phosphate transport to the lumen of the endoplasmatic reticulum where the enzymatic reaction occurs. The signs and symptoms of Type I Glycogenosis appear usually in the first year of life and include a prominent abdomen due to hepatomegaly, hypoglycemia, hyperlipidemia, hyperuricemia and hyperlactacidemia, as well as neutropenia, neutrophilic dysfunction, recurrent infections and enteritis in Type Ib Glycogenosis. Long term complications comprise hepatic adenomas and hepatocarcinoma, renal failure, gout, osteoporosis and plaquetary dysfunction. For diagnostic purposes, it is recommended DNA analysis combined with clinical and laboratory findings, and for pre-natal diagnosis amniocytes DNA study is done. Treatment is essentially aimed at avoiding metabolic changes and at reducing growth retardation and early deterioration of renal function; it consists mainly in glycemic control and can be complemented with pharmacological management. When these treatments are unsuccessful, one can consider hepatic or renal transplant.Key-words: type I glycogenosis; glucose-6-phosphatase; hypoglycemia.
INTRODUÇÃO
O primeiro relato da Glicogenose tipo I (GSD1) surgiu em 1929 por von Gierke, num artigo intitulado “Hepato-nefro-megalia-glicogénica”, onde demonstrou evidências clínicas, patológicas, microscópicas e bioquímicas de acumulação exagerada de glicogénio no tecido hepático (1). Em 1952, Gerty e CarlCori(2) estudaram a actividade da glicose-6-fosfátase (G6Pase) em homogeneizados hepáticos de doentes com glicogénio aumentado no fígado e verificaram que, em dois deles, essa actividade era extremamente baixa. Foi a primeira vez na história que se estabeleceu uma relação causal entre um defeito enzimático e uma doença metabólica congénita, no caso, a GSD1 (1,3).
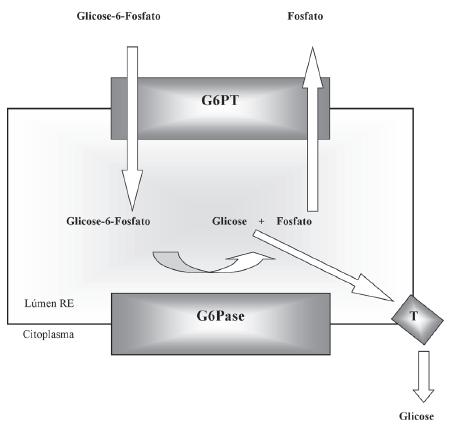
A G6Pase é uma enzima do retículo endoplasmático (RE) (4). De facto, pode ser mais adequado dizer-se que a G6Pase é um complexo funcional constituído por uma unidade catalítica com o centro activo no lúmen do RE e por transportadores que permitem quer a entrada do substrato, glicose-6-fosfato (G6P), quer a saída dos produtos da reacção, glicose e fosfato inorgânico (Pi) (Figura 1) (5-7).
Fig. 1 - Apresentação esquemática da constituição do sistema da glicose-6-fosfátase. A unidade catalítica (G6Pase), o trocador da glicose-6-fosfato/fosfato (G6PT) e o putativo transportador da glicose (T) encontram-se ancorados à membrana do retículo endoplasmático (RE).
Em 1993, a equipa liderada por Janice Chou clonou e caracterizou o gene da G6Pase humana (8), localizado no braço longo do cromossoma 17 (9), tendo ainda identificado algumas mutações causadoras da GSD1 (8,9). Assim, através da análise de DNA, passou a ser possível fazer o diagnóstico desta doença de uma maneira rápida, precisa e não invasiva e abandonar metodologias anteriores que exigiam biópsia hepática e o estudo da actividade enzimática (10).
]]> Desde o início da caracterização desta doença que vários subtipos foram sendo propostos. Com base em ensaios em microssomas hepáticos foram definidos cinco subtipos de GSD1, nomeadamente 1a, 1aSP, 1b, 1c e 1d (11).Após a descrição do primeiro subtipo, a Glicogenose tipo Ia (GSD1a), que é causado pela deficiência da unidade catalítica (2), Lange e colaboradores (12) verificaram que, em alguns doentes diagnosticados com GSD1, a G6Pase tinha uma actividade normal em tecido congelado. O trabalho experimental subsequente permitiu concluir que, nestes casos, a G6Pase está latente nos microssomas intactos e que retoma a sua actividade quando estes são permeabilizados, permitindo o contacto directo entre a enzima e o substrato (12). Ou seja, nestes casos, o problema não se encontra na unidade catalítica do sistema, mas sim no transportador (G6PT) que introduz o substrato, a G6P, no lúmen do RE (12). O gene que se encontra alterado neste subtipo, denominado Glicogenose tipo Ib (GSD1b), foi localizado na região 11q23 (13).
Em 1983, foi descrito um caso de um suposto terceiro subtipo da doença, Glicogenose tipo Ic (GSD1c); o defeito estaria no hipotético transportador responsável pelo transporte de Pi do RE para o citoplasma (14). Fenske e colaboradores (15) localizaram o gene responsável pela GSD1c no braço longo do cromossoma 11. O facto deste gene se encontrar em localização idêntica à do gene do G6PT levou outros autores a duvidarem da existência de transportadores diferentes para o G6P e para o Pi (16) e a defenderem que na prática apenas devem ser considerados dois tipos de GSD1: GSD1a e GSD1b (17). Corroborando esta ideia, Chen e colaboradores (18) descobriram recentemente (2008) que o gene do G6PT se encontra mutado nas GSD1b e GSD1c e que o G6PT funciona como um trocador, que introduz G6P no RE e retira o Pi formado. Actualmente, acredita-se que os doentes anteriormente diagnosticados com GSD1c são, na realidade, possuidores de GSD1b (11).
Apesar de se ter admitido a existência de uma alteração numa hipotética proteína estabilizadora da G6Pase que seria responsável pela Glicogenose 1aSP (19), foi demonstrado que os doentes que foram diagnosticados desta forma têm uma mutação no gene que codifica o componente catalítico, pertencendo, portanto, ao subtipo GSD1a (20). Do mesmo modo, não há evidências genéticas ou outras que permitam considerar a existência da Glicogenose 1d (GSD1d), que seria causada por deficiência na saída da glicose do RE (1). O mecanismo de transporte da glicose na membrana do RE é ainda desconhecido (21).
Apesar da vasta investigação dos últimos 80 anos, os mecanismos patogénicos que expliquem os sinais e sintomas da GSD1 são, em grande parte, ainda desconhecidos e o esclarecimento destes mecanismos, assim como a identificação de novas mutações são temas actuais de investigação. Estes aspectos serão abordados com mais pormenor ao longo desta revisão.
GENÉTICA
A GSD1 é uma doença autossómica recessiva com uma incidência estimada de 1 em cada 100.000 nascimentos (22).
De acordo com uma página da internet actualizada por Deeksha Bali e Yuan-Tsong Chen (23), estavam, à data da última actualização, descritas 86 e 80 mutações para, respectivamente, a GSD1a e GSD1b. Embora a doença não seja restrita a nenhum grupo étnico, algumas mutações foram predominantemente encontradas em determinadas populações (3,22).
Para além da heterogeneidade correspondente aos dois subtipos da doença (GSD1a e 1b), dentro de cada subtipo os doentes exibem alguma variedade fenotípica (3,22). No caso da GSD1a, embora não exista uma relação genótipo-fenótipo estrita para cada uma das mutações, algumas delas parecem conferir sintomas mais graves (22). Por exemplo, numa das mutações está mesmo descrito que os doentes podem apresentar anormalidades dos neutrófilos, situação atípica na GSD1a, mas comum na GSD1b (24).
]]>CLÍNICA E PATOGENIA
Muitas das manifestações clínicas e alterações laboratoriais são sobreponíveis nos dois subtipos da GSD1.
Os doentes são geralmente de pequena estatura e com um abdómen globoso devido à hepatomegalia. Em termos analíticos, observa-se predominantemente hipoglicemia, hiperlipidemia, hiperuricemia e hiperlactacidemia. Os indivíduos com GSD1b, a forma mais grave da doença, podem ainda apresentar neutropenia, disfunção neutrofílica, infecções recorrentes e enterite (25). Além destas manifestações que, normalmente, são aquelas a partir das quais se levanta um elevado grau de suspeição quanto à presença de GSD1, os doentes podem ter complicações crónicas, como adenomas hepáticos e hepatocarcinoma, insuficiência renal, gota, osteoporose e disfunção plaquetária (25).
A idade de apresentação deste quadro clínico/analítico é variável, podendo manifestar-se desde o primeiro dia de vida até à idade adulta (com idade média aos 6 meses) na GSD1a e do primeiro dia de vida até aos 4 anos (com idade média aos 4 meses) na GSD1b. Apesar destas diferenças, 80% dos doentes com GSD1a e 90% dos que têm GSD1b apresentam sinais e sintomas antes de completar o primeiro ano de vida (25).
Devido à introdução de técnicas terapêuticas que são eficazes na prevenção dos episódios de hipoglicemia sintomática, a esperança média de vida dos doentes com GSD1 tem aumentado nos últimos anos tendo, por isso, ganhado relevância como causa de morte as complicações tardias da doença (doença renal progressiva e as complicações dos adenomas hepáticos). Um tratamento dietético adequado pode proporcionar à maioria dos doentes adultos uma vida praticamente normal e diminuir a morbilidade durante a infância (25).
As hipóteses que procuram explicar as alterações mais relevantes da GSD1 serão discutidas de seguida.
Hipoglicemia
O passo final da gliconeogénese e da glicogenólise é a reacção G6P + H2O → Glicose + Pi, precisamente aquela que se encontra gravemente comprometida na GSD1. Estando comprometida a produção endógena de glicose no fígado e rim, a hipoglicemia é, portanto, uma complicação previsível desta doença. No entanto, muitos aspectos relacionados com a hipoglicemia e com a resposta adaptativa do organismo permanecem ainda mal esclarecidos (1).
]]> Embora se pudesse pensar que a ausência de G6Pase podia ser um impedimento absoluto à produção hepática de glicose, a verdade é que os doentes com GSD1 têm taxas de produção hepática de glicose que podem ser muito próximas do normal (26, 27). Estudos em que se usou glicose (28) ou glicerol (29) marcados permitem concluir que a glicose libertada para o sangue não provém de uma actividade residual da G6Pase. Adicionalmente, a observação de que a produção de glicose diminui com a administração de grandes quantidades de glicose enfraquece a possibilidade de que a alfa-1,4-glicosidase (maltase ácida) esteja envolvida uma vez que esta enzima é, supostamente, insensível a variações na concentração de substrato e hormonas (28). Por exclusão de partes, foi avançada a ideia de que a amilo-1,6-glicosidase (enzima desramificante) possa ter algum papel na produção endógena de glicose (28). No entanto, esta hipótese não foi apoiada por um estudo subsequente, onde se refutou experimentalmente o pressuposto de que o aumento da produção de glicose pelo fígado, verificado na ausência de administração de glicose, se deve, nestes doentes, a um aumento da velocidade do ciclo glicogénese/glico-genólise (27).Shieh e colaboradores (30) demonstraram a existência de uma proteína semelhante à G6Pase, também do RE, mas que, ao contrário desta–que é quase exclusivamente expressa no fígado, rim e intestino (21) – teria uma expressão ubiquitária. Esta proteína, a que chamaram de glicose-6-fosfátase β (G6Paseβ), teria a capacidade de se acoplar funcionalmente ao G6PT e formar um complexo G6Pase activo. Embora a G6Paseβ tenha apenas 12% da actividade da primeira G6Pase descoberta (que os autores renomearam de Glicose-6-fosfátaseα (G6Paseα)) (30), este estudo levanta importantes questões acerca da possibilidade de outros tecidos periféricos poderem participar activamente na produção endógena da glicose. Por conterem G6Paseβ e representarem uma massa substancial, os músculos poderiam ter particular importância neste contexto (31) Uma importante característica da doença, que pode estar de acordo com esta hipótese, é a verificação de que, acompanhando o aumento da massa muscular, os níveis glicémicos dos doentes aumentam desde o nascimento até à idade adulta (30).
Em 2006, Wang e colaboradores (32) criaram ratinhos transgénicos sem G6Paseβ. Em contraste com os ratinhos transgénicos sem G6Paseα e com os doentes com GSD1 observaram que, no défice de G6Paseβ, o atraso de crescimento era pouco pronunciado e que os níveis de glicogénio hepático assim como a glicemia e a trigliceridemia eram normais (32). Estes dados demonstram que o papel da G6Paseβ na produção endógena de glicose é pouco relevante quando não há defeito na G6Paseα, mas não excluem a possibilidade de ter alguma importância nos doentes com GSD1. No entanto, um dado que enfraquece esta possibilidade é o facto de os doentes com GSD1b terem uma produção endógena de glicose comparável à dos doentes com GSD1a (27), sabendo que a ausência de actividade da G6PT anula as actividades quer da G6Paseα quer da G6Paseβ; ambas as enzimas dependem da actividade da G6PT para interagirem com o substrato (30).
Os doentes com GSD1 são, relativamente aos indivíduos normais, tolerantes a baixos níveis de glicemia e os resultados de um estudo de ressonância magnética nuclear poderão ajudar a explicar este fenómeno (33). Os doentes com GSD1 têm maiores concentrações intracerebrais de glicose o que, conjuntamente com a hiperlactacidemia e a oxidação cerebraldelactato, poderia atenuar os sintomas da hipoglicemia (33).Aparentemente, a hipoglicemia pode aumentar, nas células cerebrais, a capacidade de captação da glicose circulante (33).
Hiperlactacidemia
Na génese da hiperlactacidemia está o aumento da produção hepática de lactato devido à acumulação de G6P intra-hepatocitária (33) e consequente aumento da sua utilização na via da glicólise (com maior produção de piruvato e, consequentemente, de lactato) assim como a sua menor utilização na gliconeogénese (29). Embora a conversão de piruvato em acetil-CoA e a de fosfoenolpiruvato em G6P estejam aumentadas nos doentes com GSD1, o principal produto formado a partir dos substratos da gliconeogénese é o lactato (29). Nos doentes com GSD1, a hiperlactacidemia diminui quando se administra glicose e os níveis glicémicos sobem (34). Pode-se especular que na génese deste fenómeno esteja a consequente subida da insulina que, levando à activação da desidrogénase do piruvato (35), provoque um desvio de fluxo com um relativo aumento da oxidação do lactato a piruvato e deste a acetil-CoA.
Hiperuricemia
O aumento da degradação do ATP em resposta à hipoglicemia foi proposto como um dos mecanismos para a génese da hiperuricemia (36). Greene e colaboradores (36) usaram a infusão de glicagina para simular as alterações bioquímicas provocadas pela hipoglicemia, nomeadamente a activação da fosforílase do glicogénio hepática. Nos seus estudos observaram que a infusão de glicagina em doentes com GSD1 aumentava os níveis intra-hepáticos de G6P, frutose-6-fosfato (F6P) e frutose-1,6-bifosfato (F1,6BP) e causava uma redução marcada nos níveis de ATP (36). Esta alteração pode dever-se ao facto de que a glicagina, aumentando os níveis de G6P e F6P aumentaria o gasto de ATP no passo da fosforilação da F6P a F1,6BP. Esta depleção de ATP levaria a um aumento secundário da concentração intracelular e da degradação do AMP, com a consequente formação de ácido úrico. Também se verificou que a prevenção da hipoglicemia reduzia a uricemia, devido à consequente diminuição dos níveis de lactato (36), visto este ser um inibidor da excreção renal de urato.
]]>Hiperlipidemia, Esteatose hepática e Aterosclerose
A hepatomegalia acentuada dos indivíduos com GSD1 é devida quer à deposição lipídica, quer à acumulação do glicogénio (37, 38). Os doentes com GSD1 têm níveis baixos de insulina, o que explicaria o aumento da concentração de ácidos gordos livres plasmáticos em consequência da estimulação da lipólise no tecido adiposo (38). A esteatose hepática seria uma consequência da captação pelo fígado destes ácidos gordos e da sua subsequente esterificação a triglicerídeos e ésteres de colesterol (38).
Os níveis séricos de triglicerídeos e colesterol podem chegar a valores muito altos: na ordem de 6000 mg/dl e 600 mg/dl, respectivamente (39).
Nestes doentes, as VLDL e LDL estão não só aumentadas em número, como em tamanho, devido a uma maior acumulação de triglicerídeos (40). Esta hiperlipidemia pode ser em parte causada pelo aumento da formação de acetil-CoA (29), que é substrato na síntese de colesterol e ácidos gordos. Além disso, alguns intermediários do metabolismo da glicose cuja concentração intracelular aumenta na GSD1 (como a G6P ou a xilulose-5-fosfato, que está em equilíbrio com a G6P (41)) são estimuladores da lipogénese (38, 42).
A insulinainibe a secreção de VLDL e estimula alípase delipoproteínas do tecido adiposo; dada a hipoinsulinemia crónica nos doentes com GSD1 é, por isso, de esperar que a secreção hepática de VLDL esteja aumentada (38). No entanto, o aumento esperado da secreção destas partículas não foi observado num estudo em ratos onde se inibiu farmacologicamente o G6PT (43). No caso das LDL, uma explicação para o seu aumento seria a diminuição da sua captação pelos tecidos (38).
Um aspecto interessante é a verificação de que, apesar da dislipidemia marcada, os doentes não têm aterosclerose prematura e os vários estudos não apoiam o uso de terapêutica hipolipidémica para a sua prevenção (44). A desintoxicação dos radicais livres pode ser um factor nesta protecção (45) e o aumento do urato plasmático, um factor anti-oxidante, pode ter, aqui, um papel relevante (46).
Disfunção plaquetária
Outra explicação para a baixa incidência de aterosclerose é a diminuição da aderência plaquetária verificada nos doentes com GSD1. Uma vez que a G6Pase não existe nas plaquetas, esta disfunção não é secundária a anomalias desta enzima na plaqueta (47).
]]> Com base na observação de que a terapêutica dietética correctora da hipoglicemia corrigia a disfunção plaquetária, Hutton e colaboradores (48) sugeriram que a hipoglicemia per se seria um factor etiológico nesta disfunção. A hipoglicemia crónica poderá implicar níveis reduzidos de glicose na plaqueta e, consequentemente, uma diminuição da via glicolítica, com menor formação de ATP; a razão ATP/ADP é normal, o que, a par com a presumível descida da concentração de ATP, pode reflectir uma diminuição geral dos nucleotídeos de adenina plaquetários (48). Sabendo-se que estes nucleotídeos são importantes para a função e agregação das plaquetas, esta poderá ser uma explicação para a disfunção plaquetária dos doentes com GSD1.Mais recentemente (2005), verificou-se que 60% dos doentes com GSD1 tinham níveis moderadamente baixos do factor de von Willebrand que, embora raramente contribuíssem para uma maior tendência hemorrágica, poderiam contribuir para a protecção contra a aterosclerose (49).
Disfunção dos neutrófilos/neutropenia
Devido às complicações infecciosas graves que frequentemente acometem os doentes com GSD1b, estes têm geralmente um prognóstico pior que o dos doentes com GSD1a (50).
A neutropenia é frequentemente encontrada na GSD1b: verificou-se que 87% destes doentes apresentam neutropenia no decorrer da doença, sendo intermitente na maioria dos casos (50). Além de neutropenia há disfunção dos neutrófilos e quer esta disfunção, quer a neutropenia manifestam-se, geralmente, no primeiro ano de vida (50). Os defeitos dos neutrófilos incluem menor burst respiratório (reacção verificada na activação dos neutrófilos e na fagocitose, com consumo de O2e produção de superóxido por acção da oxídase do NADPH) assim como alterações na quimiotaxia, fagocitose e sinalização pelo cálcio (50).
A disfunção dos neutrófilos e a neutropenia tornam estes doentes susceptíveis a estomatite aftosa, doença inflamatória intestinal e infecções bacterianas recorrentes (39).
Uma explicação para a disfunção dos neutrófilos foi encontrada quando se mostrou que uma série de sinais de apoptose estavam anormalmente presentes nos neutrófilos dos doentes com GSD1b (51). Embora os neutrófilos normais não expressem a G6Paseα, expressam o G6PT (52). Estando esta última proteína em défice na GSD1b é de prever que a função de transporte da G6P (ou do Pi) no RE dos neutrófilos possa estar na origem da apoptose observada.
De facto, Leuzzi e colaboradores (53) demonstraram que a inibição farmacológica do G6PT em neutrófilos isolados provocava uma menor produção de anião superóxido, uma menor concentração de cálcio no RE e um aumento da proporção de células apoptóticas, sendo estes resultados compatíveis com os achados nos neutrófilos dos doentes com GSD1b (51). Uma vez que a apoptose dependente desta inibição foi prevenida por tratamento que diminuiu o stress oxidativo admite-se a hipótese de que o defeito no transporte da G6P presente nos doentes com GSD1b resultaria numa diminuição das defesas antioxidantes (53). De facto, a síntese de NADPH no lúmen do RE está dependente da presença de G6P, que é substrato da desidrogénase da hexose-6-fosfato do RE (53).
Um outro possível destino da G6P que entrou para o RE dos neutrófilos (via G6PT) é a sua hidrólise por acção da G6Paseβ (52). Num estudo de Cheung e colaboradores (52) com ratinhos que não expressam esta enzima, mostrou-se que estes ratinhos não exibem distúrbios da homeostasia da glicose mas apresentam maior susceptibilidade a infecções assim como neutropenia e disfunções nos neutrófilos (incluindo apoptose) semelhantes às exibidas por ratinhos deficientes em G6PT e pelos indivíduos com GSD1b. Este estudo também apoia a especulação de que, na origem das alterações dos neutrófilos na GSD1b poderia estar a deficiência na actividade do complexo funcional G6Paseβ-G6PT nos neutrófilos com a consequente diminuição da produção intraluminal de glicose no RE e indução de apoptose (52).
]]> Um estudo de Kim e colaboradores (54) em ratinhos que não expressam G6Paseα mostrou que, pelo menos em parte, as alterações da homeostasia da glicose per se podem ter algum papel na disfunção mielóide, estando esta função mais alterada na GSD1b, por se apresentar em conjunto com o défice de G6PT. Em consonância com a normal ausência de G6Paseα nos neutrófilos e indicando que a G6Paseα não é necessária para a função dos neutrófilos, os neutrófilos desses ratinhos não apresentavam defeitos no burst respiratório, na quimiotaxia ou na sinalização do cálcio. Contudo, embora em grau menos marcado que nos ratinhos que não expressam a G6PT, apresentavam outras alterações como níveis séricos elevados de factor estimulante de colónias de granulócitos (granulocyte colony stimulating factor (G-CSF)) e quimioatractor de neutrófilos induzido por citocinas (cytokine-induced neutrophil chemoattractant (KC)), aumento das células progenitoras mielóides na medula óssea e no baço e hipocelularidade e atraso no desenvolvimento do osso e do baço. Em consonância com os níveis elevados de G-CSF e KC, os ratinhos em análise apresentavam neutrofilia (54).
Alterações do crescimento
Em 2003, Mundy e colaboradores (55) estudaram as variáveis metabólicas que podem afectar o crescimento dos doentes com GSD1. Um dado relevante foi o que resultou da comparação dos doentes deste estudo com o de um outro que teve lugar, na mesma instituição, em 1982 (56), ou seja, antes da introdução da terapêutica dietética intensiva que previne os episódios de hipoglicemia sintomática. Embora os doentes do estudo de 2003 fossem baixos relativamente aos controlos, eram significativamente mais altos que os do estudo de 1982.
De acordo com o estudo em análise (55), um distúrbio no eixo Hormona de Crescimento (HC) – factor de crescimento semelhante à insulina tipo 1 (insuline-like growth factor 1 (IGF-1)) é o responsável pelas alterações do crescimento. As crianças pré-púberes com esta doença tinham periodicidade secretória de HC normal, mas os picos e os valores médios eram inferiores aos de crianças saudáveis de baixa estatura. Além disso, entre as crianças doentes estudadas, as de menor estatura apresentaram maior insensibilidade à HC e menores níveis de IGF-1 (55). Esta insensibilidade pode dever-se aos elevados níveis de cortisol observados nos doentes de menor estatura, o que está de acordo com o efeito desta hormona que antagoniza o aumento do IGF-1 RNAm induzido pela HC (55). Os doentes com GSD1 têm, devido à hiperlactacidemia, acidose metabólica crónica, que também foi proposta como responsável pela insensibilidade à HC (57).
Os doentes de menor estatura apresentavam os valores mais altos de HC; parece, por isso, pouco provável que o tratamento com HC exógena consiga compensar a resistência hepática e ter assim algum êxito terapêutico (55).
Adenomas hepáticos e hepatocarcinoma
A prevalência de adenomas hepáticos nos doentes com GSD1 é de 22 a 75%, sendo maior em idades avançadas e semelhante nos dois sexos (58). Estes adenomas podem ser solitários ou múltiplos e podem complicar-se com hemorragia ou transformação maligna (58).
Uma hipótese avançada para explicar o aparecimento de adenomas relaciona-se com as alterações no metabolismolipídico. Na GSD1 há uma maior libertação de ácidos gordos livres pelo tecido adiposo; é possível especular que pelo menos alguns dos ácidos gordos que chegam ao fígado possam ser oxidados nos peroxissomas, levando à produção de H2O2 que pode estar envolvido na origem de mutações no DNA e na génese dos tumores (58).
]]> Um estudo recente (2008) não detectou diferenças nos parâmetros bioquímicos e no tratamento dietético em doentes com GSD1 que desenvolveram adenomas relativamente aos que não desenvolveram (59). O risco de transformação maligna dos adenomas também é incerto e, por isso, o aconselhamento dos doentes é problemático, podendo-se usar ecografia hepática para a detecção e seguimento dos tumores (58). O transplante hepático é o tratamento definitivo (58).
Insuficiência renal
A G6Paseα existe normalmente nas células tubulares renais e os doentes com GSD1 têm glicogénio aumentado nestas células e nefromegalia (21, 60). O défice de produção de glicose no rim pode contribuir para a hipoglicemia mas as relações etiopatogénicas entre o défice de G6Pase nas células tubulares renais e a nefropatia dos doentes com GSD1 permanecem mal esclarecidas (61).
Num estudo de Baker e colaboradores (60) a taxa de filtração glomerular (TFG) estava aumentada em praticamente todos os doentes com GSD1 observados. Embora esta possa ser a única alteração verificada nos doentes mais jovens, um aumento significativo da excreção urinária de albumina foi observado nos doentes adolescentes, podendo evoluir para uma proteinúria marcada, inclusivamente para níveis nefróticos (60). Não foram observadas quaisquer relações entre a TFG e o aporte proteico, a altura e o peso, a pressão arterial, os níveis lipídicos ou a existência/ausência de terapêutica com alimentação contínua (60). Para além das alterações acima referidas, os doentes com GSD1 também podem ter hipertensão e hematúria (62). As biópsias renais, além da já referida acumulação tubular de glicogénio, mostraram que havia fibrose intersticial e glomeruloesclerose segmentar focal/global (60). A insuficiência renal requerendo hemodiálise ou transplante é uma das evoluções possíveis da nefropatia (25).
Um estudo recente (2008) com ratinhos transgénicos que não exprimem a G6Paseα mostrou que um dos factores envolvidos na etiologia da nefropatia dos doentes com GSD1 poderá ser a expressão aumentada de angiotensinogénio nas células tubulares renais proximais e o consequente aumento da síntese de factores pró-fibróticos dependentes da angiotensina II (61). Nestes ratinhos havia aumento da síntese de colagénio e fibronectina, que são proteínas da matriz extracelular; tal como os doentes com GSD1, os ratinhos apresentavam fibrose intersticial renal e glomeruloesclerose (61).
DIAGNÓSTICO
Actualmente, o diagnóstico da GSD1 consiste na análise de mutações combinada com as alterações clínicas e bioquímicas (63).
Para a identificação das mutações dos genes da G6Pase e do G6PT, a técnica a usar pode ser dirigida às mutações específicas mais comuns, como por exemplo, PCR-RFLP (64) e, no caso de o resultado ser negativo, faz-se a sequenciação directa do gene.
]]> Já em 2000, Rake e colaboradores (65) defendiam a utilização da análise de mutações combinada com as alterações clínicas e bioquímicas e o abandono dos estudos enzimáticos efectuados em biópsias de tecido hepático. Só nos raros casos em que o diagnóstico genético não for possível ou inconclusivo e se a suspeição de GSD1 se mantiver é que poderá ser feito um estudo enzimático em tecido hepático, sendo necessária a medição da actividade da G6Pase em microssomas intactos e em microssomas permeabilizados (65).O diagnóstico pré-natal também é possível, através do estudo do DNA de amniócitos.
TRATAMENTO
Os objectivos do tratamento são prevenir alterações metabólicas agudas, nomeadamente os episódios de hipoglicemia sintomática, e proporcionar desenvolvimento psicomotor normal assim como uma boa qualidade de vida (63). Com os métodos terapêuticos adequados a esperança média de vida ultrapassa a terceira década (39).
Não há cura para a GSD1 (3). O controlo glicémico constitui a base do tratamento da GSD1; quando a hipoglicemia é prevenida, as alterações bioquímicas e o atraso no desenvolvimento são atenuados (66, 67); alguns autores admitem também que o aparecimento dos adenomas hepáticos e da lesão renal podem ser retardados (67). De maneira a manter concentrações de glicose acima do limiar desencadeador de sintomatologia e de outras alterações metabólicas, a glicose pode ser administrada continuamente por infusão intragástrica (via tubo nasogástrico ou gastrostomia) ou pelo uso de alimentos com baixo índice glicémico, como o amido de milho não cozinhado (AMNC) (66).
Embora os ensaios ainda sejam limitados a ratinhos, a terapêutica genética usando adenovírus parece ser promissora (68, 69).
O transplante hepático deve ser considerado quando o tratamento dietético falha, ou quando há irressecabilidade ou suspeita de transformação maligna de um adenoma hepático (70). O transplante hepático normaliza os parâmetros metabólicos assim como o crescimento físico e intelectual (71). Também foi proposto transplante de rim (70) ou transplante combinado fígado/rim em doentes com insuficiência renal terminal, mesmo na ausência de lesões tumorais hepáticas (72).
Pierre e colaboradores (73) descrevem um caso de sucesso com transplante de medula óssea, defendendo que é uma opção de tratamento viável para doentes com GSD1b com doença inflamatória intestinal e infecções recorrentes.
Embora não evite a apoptose precoce dos neutrófilos (51), a administração de factor estimulador das colónias granulocíticas parece ser uma terapêutica promissora para os doentes com GSD1b; este tratamento induz aumento do número de neutrófilos e redução dos sintomas da doença inflamatória intestinal (39). No entanto, pode haver um aumento do risco de leucemia mielóide aguda e síndromes mielodisplásicos com este tratamento; as análises recorrentes à medula óssea são por isso recomendadas (74).
]]> Outras terapêuticas farmacológicas a considerar são, por exemplo, o alopurinol para a hiperuricemia, o bicarbonato para a acidemia, os inibidores da enzima de conversão da angiotensina para retardar a progressão da lesão renal e suplementos vitamínicos.
PERSPECTIVAS FUTURAS
Apesar do primeiro caso de GSD1 ter sido descrito há já 80 anos, torna-se evidente que se desconhece a génese de muitas alterações desta patologia e o seu relacionamento com o defeito primário (na G6Pase ou no G6PT). A investigação prossegue com o objectivo de prevenir as complicações e descobrir quais as terapêuticas mais apropriadas para a melhoria da qualidade de vida e que minimizem as sequelas dos defeitos metabólicos.
Agradecimentos
Os autores agradecem à direcção do Serviço de Bioquímica da Faculdade de Medicina da Universidade do Porto e à FCT (POCI, Feder e Programa Comunitário de Apoio). Os autores desejam ainda prestar a merecida homenagem à Professora Isabel Azevedo que, após uma vida dedicada à Faculdade de Medicina e à Universidade do Porto, pediu recentemente a reforma abandonado assim as funções de Directora do Serviço de Bioquímica que desempenhou com amor e superior competência.
REFERÊNCIAS
1 -Moses SW. Historical highlights and unsolved problems in glycogen storage disease type 1. Eur J Pediatr 2002;161: S2-9.
[ Links ]2 -Cori GT, Cori CF. Glucose-6-phosphatase of the liver in glycogen storage disease. J Biol Chem 1952;199:661-7.
3 -Chou JY, Matern D, Mansfield BC, Chen YT. Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med 2002;2:121-43.
4 -Hers HG, Berthet J, Berthet L, De Duve C. [The hexose-phosphatase system. III. Intracellularlocalization of enzymes by fractional centrifugation.]. Bull Soc Chim Biol (Paris) 1951;33:21-41.
5 -Arion WJ, Wallin BK, Lange AJ, Ballas LM. On the involvement of a glucose 6-phosphate transport system in the function of microsomal glucose 6-phosphatase. Mol Cell Biochem 1975;6:75-83.
6 -Arion WJ, Ballas LM, LangeAJ, Wallin BK. Microsomalmembrane permeability and the hepatic glucose-6-phosphatase system. Interactions of the system with D-mannose 6-phos-phate and D-mannose. J Biol Chem 1976;251:4891-7.
7 - Marcolongo P, Banhegyi G, Benedetti A, Hinds CJ, Burchell A. Liver microsomal transport of glucose-6-phosphate, glucose, and phosphate in type 1 glycogen storage disease. J Clin Endocrinol Metab 1998;83:224-9.
8 -Lei KJ, Shelly LL, Pan CJ, Sidbury JB, Chou JY. Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a. Science 1993;262:580-3.
9 - Lei KJ, Pan CJ, Shelly LL, Liu JL, Chou JY. Identification of mutations in the gene for glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. J Clin Invest 1994;93:1994-9.
10 -Lei KJ, Chen YT, Chen H, et al. Genetic basis of glycogen storage disease type 1a: prevalent mutations at the glucose-6-phosphatase locus. Am J Hum Genet 1995;57:766-71.
11 -Veiga-da-Cunha M, Gerin I, Van Schaftingen E. How many forms of glycogen storage disease type I? Eur J Pediatr 2000;159:314-8.
]]> 12 -Lange AJ, Arion WJ, Beaudet AL. Type Ib glycogen storage disease is caused by a defect in the glucose-6-phosphate translocase of the microsomal glucose-6-phosphatase system. J Biol Chem 1980;255:8381-4.13 -Annabi B, Hiraiwa H, Mansfield BC, Lei KJ, Ubagai T, Polymeropoulos MH, et al. The gene for glycogen-storage disease type 1b maps to chromosome 11q23. Am J Hum Genet 1998;62:400-5.
14 -Nordlie RC, Sukalski KA, Munoz JM, Baldwin JJ. Type Ic, a novel glycogenosis. Underlying mechanism. J Biol Chem 1983;258:9739-44.
15 -Fenske CD, Jeffery S, Weber JL, Houlston RS, Leonard JV, Lee PJ. Localisation of the gene for glycogen storage disease type 1c by homozygosity mapping to 11q. J Med Genet 1998;35:269-72.
16 -Veiga-da-Cunha M, Gerin I, Chen YT, et al. A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic. Am J Hum Genet 1998;63:976-83.
17 -Veiga-da-Cunha M, Gerin I, Chen YT, Lee PJ, Leonard JV, Maire I, et al. The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur J Hum Genet 1999;7:717-23.
18 -Chen SY, Pan CJ, Nandigama K, Mansfield BC, Ambudkar SV, Chou JY. The glucose-6-phosphate transporter is a phosphate-linked antiporter deficient in glycogen storage disease type Ib and Ic. FASEB J 2008;22:2206-13.
19 -Burchell A, Waddell ID. Diagnosis of a novel glycogen storage disease: type 1aSP. J Inherit Metab Dis 1990;13:247-9.
20 -Lei KJ, Shelly LL, Lin B, et al. Mutations in the glucose-6-phosphatase gene are associated with glycogen storage disease types 1a and 1aSP but not 1b and 1c. J Clin Invest 1995;95:234-40.
21 -van Schaftingen E, Gerin I. The glucose-6-phosphatase system. Biochem J 2002;362:513-32.
]]> 22 -Chou JY, Mansfield BC. Mutations in the glucose-6-phos-phatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat 2008;29:921-30.23 -Bali DS, Chen Y-T. Glycogen Storage Disease Type I. Disponível em: URL: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=gsd1, acedido em 11 de Setembro de 2009.
24 -Weston BW,Lin JL,Muenzer J,et al.Glucose-6-phosphatase mutation G188R confers an atypical glycogen storage disease type 1b phenotype. Pediatr Res 2000;48:329-34.
25 -Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinicalcourse and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 2002;161:S20-34.
26 -Powell RC, Wentworth SM, Brandt IK. Endogenous glucose production in Type I glycogen storage disease. Metabolism 1981;30:443-50.
27 -Rother KI, Schwenk WF. Glucose production in glycogen storage disease I is not associated with increased cycling through hepatic glycogen. Am J Physiol 1995;269:E774-8.
28 -Kalderon B, Korman SH, Gutman A, Lapidot A. Estimation of glucose carbon recycling in children with glycogen storage disease: A 13C NMR study using [U-13C]glucose. Proc Natl Acad Sci U S A 1989;86:4690-4.
29 -Jones JG., Garcia P, Barosa C, Delgado TC, Diogo L. Hepatic anaplerotic outflow fluxes are redirected from gluconeogenesis to lactate synthesis in patients with Type 1a glycogen storage disease. Metab Eng 2009;11:155-62.
30 -Shieh JJ, Pan CJ, Mansfield BC, Chou JY. A glucose-6-phosphate hydrolase, widely expressed outside the liver, can explain age-dependent resolution of hypoglycemia in glycogen storage disease type Ia. J Biol Chem 2003;278:47098-103.
31 -Shieh JJ, Pan CJ, Mansfield BC, Chou JY. A potential new role for muscle in blood glucose homeostasis. J Biol Chem 2004;279:26215-9.
]]> 32 -Wang Y, Oeser JK, Yang C, et al. Deletion of the gene encoding the ubiquitously expressed glucose-6-phospha-tase catalytic subunit-related protein (UGRP)/glucose-6-phosphatase catalytic subunit-beta results in lowered plasma cholesterol and elevated glucagon. J Biol Chem 2006;281:39982-9.33 -Weghuber D, Mandl M, Krssak M, et al. Characterization of hepatic and brain metabolism in young adults with glycogen storage disease type 1: a magnetic resonance spectroscopy study. Am J Physiol Endocrinol Metab 2007;293:E1378-84.
34 -Stanley CA, Mills JL, Baker L. Intragastric feeding in type I glycogen storage disease: factors affecting the control of lactic acidemia. Pediatr Res 1981;15:1504-8.
35 -Sugden MC, Holness MJ. Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 2006;112:139-49.
36 -Greene HL, Wilson FA, Hefferan P, et al. ATP depletion, a possible role in the pathogenesis of hyperuricemia in glycogen storage disease type I. J Clin Invest 1978;62:321-8.
37 -Hers HG, Van-Hoof F, de Barsy T. Glycogen storage diseases. In: Scriver CR, BeaudetAL, Sly WS, Valle D, editors. The metabolic basis of inherited disease. 6th ed. New York: McGraw-Hill Information Services Co; 1989. p. 425-52
38 -Bandsma RH, Smit GP, Kuipers F. Disturbed lipid metabolism in glycogen storage disease type 1. Eur J Pediatr 2002;161:S65-9.
39 -Reis CV, Penna FJ, Oliveira MC, Roquete ML. Glicogenose tipo I. J Pediatr (Rio J) 1999;75:227-36.
40 -Levy E, Thibault LA, Roy CC, Bendayan M, Lepage G, Letarte J. Circulating lipids and lipoproteins in glycogen storage disease type I with nocturnal intragastric feeding. J Lipid Res 1988;29:215-26.
41 -Veech RL. A humble hexose monophosphate pathway metabolite regulates short-andlong-term controloflipogenesis. Proc Natl Acad Sci U S A 2003;100:5578-80.
]]> 42 -Bandsma RH, Prinsen BH, van Der Velden Mde S, Rake JP, Boer T, Smit GP, et al. Increased de novo lipogenesis and delayed conversion of large VLDLinto intermediate density lipoprotein particles contribute to hyperlipidemiain glycogen storage disease type 1a. Pediatr Res 2008;63:702-7.43 -Bandsma RH, Wiegman CH, Herling AW, et al. Acute inhibition of glucose-6-phosphate translocator activity leads to increased de novo lipogenesis and development of hepatic steatosis without affecting VLDL production in rats. Diabetes 2001;50:2591-7.
44 -bels FL, Rake JP, Slaets JP, Smit GP, Smit AJ. Is glycogen storage disease 1a associated with atherosclerosis? Eur J Pediatr 2002;161:S62-4.
45 -Wittenstein B, Klein M, Finckh B, Ullrich K, Kohlschutter A. Plasma antioxidants in pediatric patients with glycogen storage disease, diabetes mellitus, and hypercholesterolemia. Free Radic Biol Med 2002;33:103-10
46 -Wittenstein B, Klein M, Finckh B, Ullrich K, Kohlschutter A. Radical trapping in glycogen storage disease 1a. Eur J Pediatr 2002;161:S70-4.
47 -Czapek EE, Deykin D, Salzman EW. Platelet dysfunction in glycogen storage disease type I. Blood 1973;41:235-47.
48 -Hutton RA, Macnab AJ, Rivers RP. Defect of platelet function associated with chronic hypoglycaemia. Arch Dis Child 1976;51:49-55.
49 -Muhlhausen C, Schneppenheim R, Budde U, et al. Decreased plasma concentration of von Willebrand factor antigen (VWF:Ag)in patients with glycogen storage disease type Ia. J Inherit Metab Dis 2005;28:945-50.
50 -Visser G, Rake JP, Fernandes J, et al. Neutropenia, neutrophil dysfunction, and inflammatory bowel disease in glycogen storage disease type Ib: results of the European Study on Glycogen Storage Disease type I. J Pediatr 2000;137:187-91.
51 -Kuijpers TW, Maianski NA, Tool AT, et al. Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (GSD1b). Blood 2003;101:5021-4.
]]> 52 -Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, et al. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phospha-tase-beta. J Clin Invest 2007;117:784-93.53 -Leuzzi R, Banhegyi G, Kardon T, et al. Inhibition of micro-somalglucose-6-phosphate transportin human neutrophils results in apoptosis: a potential explanation for neutrophil dysfunction in glycogen storage disease type 1b. Blood 2003;101:2381-7.
54 -Kim SY, Chen LY, Yiu WH, Weinstein DA, Chou JY. Neutrophilia and elevated serum cytokines are implicated in glycogen storage disease type Ia. FEBS Lett 2007;581:3833-8.
55 -Mundy HR, Hindmarsh PC, Matthews DR, Leonard JV, Lee PJ. The regulation of growth in glycogen storage disease type 1. Clin Endocrinol (Oxf) 2003;58:332-9.
56 -Dunger DB, Holder AT, Leonard JV, Okae J, Preece MA. Growth and endocrine changesin the hepatic glycogenoses. Eur J Pediatr 1982;138:226-30.
57 -Brungger M, Hulter HN, Krapf R. Effect of chronic metabolic acidosis on the growth hormone/IGF-1 endocrine axis: new cause of growth hormone insensitivity in humans. Kidney Int 1997;51:216-21.
58 -Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr 2002;161:S46-9.
59 -DiRocco M, Calevo MG, Taro M, Melis D,AllegriAE, Parenti G. Hepatocellular adenoma and metabolic balance in patients with type Ia glycogen storage disease. Mol Genet Metab 2008;93:398-402.
60 -Baker L, Dahlem S, Goldfarb S, et al. Hyperfiltration and renal disease in glycogen storage disease, type I. Kidney Int 1989;35:1345-50.
61 -Yiu WH, Pan CJ, Ruef RA, et al.Angiotensin mediates renal fibrosis in the nephropathy of glycogen storage disease type Ia. Kidney Int 2008;73:716-23.
]]> 62 -Chen YT, Coleman RA, Scheinman JI, Kolbeck PC, Sidbury JB. Renal disease in type I glycogen storage disease. N Engl J Med 1988;318:7-11.63 -Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Guidelines for management of glycogen storage disease type I-European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 2002;161:S112-9.
64 -Trioche P, Francoual J, Audibert F, et al. Prenatal diagnosis of glycogen storage disease type Ia by restriction enzyme digestion. Prenat Diagn 1998;18:629-31.
65 -Rake JP, ten Berge AM, Visser G, et al. Glycogen storage disease type Ia: recent experience with mutation analysis, a summary of mutations reported in the literature and a newly developed diagnostic flow chart. Eur J Pediatr 2000;159:322-30.
66 -Weinstein DA, Wolfsdorf JI. Effect of continuous glucose therapy with uncooked cornstarch on the long-term clinical course of type 1a glycogen storage disease. Eur J Pediatr 2002;161:S35-9.
67 -Daublin G, Schwahn B, Wendel U. Type I glycogen storage disease: favourable outcome on a strict management regimen avoidingincreasedlactate production during childhood and adolescence. Eur J Pediatr 2002;161:S40-5.
68 -Chou JY, Mansfield BC. Gene therapy for type I glycogen storage diseases. Curr Gene Ther 2007;7:79-88.
69 -Yiu WH, Pan CJ, Allamarvdasht M, Kim SY, Chou JY. Glucose-6-phosphate transporter gene therapy corrects metabolic and myeloid abnormalities in glycogen storage disease type Ib mice. Gene Ther 2007;14:219-26.
70 -Labrune P. Glycogen storage disease type I: indications for liver and/or kidney transplantation. Eur J Pediatr 2002;161:S53-5.
71 -Iyer SG, Chen CL, Wang CC, et al. Long-term results of living donor liver transplantation for glycogen storage disorders in children. Liver Transpl 2007;13:848-52.
]]> 72 -Belingheri M, Ghio L, Sala A, et al. Combined liver-kidney transplantation in glycogen storage disease Ia: a case beyond the guidelines. Liver Transpl 2007;13:762-4.73 -Pierre G, Chakupurakal G, McKiernan P, Hendriksz C, Lawson S, Chakrapani A. Bone marrow transplantation in glycogen storage disease type 1b. J Pediatr 2008;152:286-8.
74 - Schroeder T, Hildebrandt B, Mayatepek E, Germing U, Haas R.Apatient with glycogen storage disease type Ib presenting with acute myeloid leukemia (AML) bearing monosomy 7 and translocation t(3;8)(q26;q24) after 14 years of treatment with granulocyte colony-stimulating factor (G-CSF): A case report. J Med Case Reports 2008;2:319.
Correspondência:
Dr. Rui Fontes
Serviço de Bioquímica
Faculdade de Medicina da Universidade do Porto
Alameda Prof. Hernâni Monteiro
4200-319 Porto
]]> e-mail: ruifonte@med.up.pt ]]>