Ana Farinha
Department of Nephrology, Centro Hospitalar de Setúbal. Setúbal, Portugal.
ABSTRACT
Atypical haemolytic uraemic syndrome is a rare disease characterised by microangiopathic haemolytic anaemia, thrombocytopaenia and predominant renal impairment in the absence of Shiga toxin-producing bacteria. For long time it has been difficult to distinguish it from other thrombotic microangiopathies, but in the last decade advances have been made in understanding the pathogenesis of atypical haemolytic uraemic syndrome as a disorder of alternative pathway of the complement system. Knowledge of mutations and polymorphisms in the genes encoding the complement regulatory proteins revealed clinical importance in the management of the patients, altering not only the transplantation perspective but also leading to the search for new drugs, something that will potentially change the poor prognosis of these patients.
]]> This article reviews the differential diagnosis of this thrombotic microangiopathy to reflect on current treatment options and discuss new therapies.Key-Words: Alternative complement pathway; atypical haemolyticuraemic syndrome; eculizumab; plasmapheresis; transplantation.
INTRODUCTION
The term thrombotic microangiopathy (TMA) has been used to describe a histopathological entity characterised by the presence of fibrin and/or platelet thrombi in the microcirculation of various organs1.
It includes two main syndromes, the haemolytic uraemic syndrome (HUS) and thrombotic thrombocytopaenic purpura (TTP). They both present clinically with microangiopathic haemolytic anaemia and thrombocytopaenia and histologically with vascular abnormalities, namely glomerular endothelial damage, swelling of the endothelium, endothelial detachment of the basement membrane, intima fibrosis and thrombosis. For a long time they were only distinguished by clinical aspects: HUS characterised by predominant renal involvement, i.e. acute renal failure, and TTP by predominant neurological involvement. ]]>
In many patients, however, the clinical presentation overlaps, making a precise diagnosis impossible.
In the last few years, two important turning points have allowed the distinction between these two entities.
The first was the identification that ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 repeats) deficiency is more likely to present with the insidious or fluctuating neurological signs of adult idiopathic TTP2-5. The second was the finding that abnormal control of the alternative complement pathway is a risk factor for atypical HUS (aHUS)6,7.
Since then, the discrimination between these two TMA is made by measuring ADAMTS13 activity, which is greatly reduced (5 to 10% of normal) in TTP. This is mostly due to autoantibodies against ADAMTS13 because congenital TTP, caused by mutations in the ADAMTS13 gene, is an extremely rare autosomal recessive disease (incidence 1:1,000,000)8.
HAEMOLYTIC-URAEMIC SYNDROME
]]> It is important to distinguish the disease triggered by an infection with Shiga-like toxin producing Escherichia coli (STEC), which accounts for 90%, from those which are not, as management and prognosis of these patients is completely different. STEC-HUS, also called typical or diarrhoea-associated HUS (D+HUS), is a disease especially affecting young children between two and six years old who are infected by enterohaemorragic Escherichia coli serotype 0157:H7 or in some tropical regions Shigella dysenteriae type 1. Three to eight days after contamination, the patient develops abdominal pain with watery and/or bloody diarrhoea, followed within 24 hours by haemolytic anaemia, thrombocytopaenia and acute renal failure.In 2011 the worlds largest STEC outbreak occurred in Germany, affecting mostly adults above 20 years old and predominantly females. It was attributed to changes in the microbial characteristics of the bacteria (STEC O104:H4)9, indicating that changes in the bacterial characteristics can lead to changes in host profile.
STEC-HUS resolves spontaneously, and no other intervention such as plasmapheresis has shown to be superior to supportive management of renal failure, anaemia, hypertension and fluid-electrolyte imbalance10.
Mortality in children with STEC-HUS is 3 to 5% during the acute phase of the disease11; about 75% of the patients recover completely after a STEC-HUS episode12. In the German outbreak, although a greater proportion of patients infected with STEC O104:H4 eventually developed HUS9, both clinical course of individual patients and mortality (~4%) seemed to be comparable with historic reports13.
When STEC-HUS progresses to endstage renal disease (ESRD), kidney transplantation is an option, with no recurrence of the disease14.
Non-STEC-HUS or aHUS is only seen in 5 to 10% of all HUS cases15. It differs from STEC-HUS in that it can appear at any age; patients have a poor prognosis with a high mortality and morbidity in the acute phase of the disease; and progression to ESRD occurs in 50% of cases16,17.
Many causes of aHUS have been identified, such as associations with non-enteric infections (especially Streptococcus pneumoniae infections, called neuroaminidase associated-HUS), viruses, malignancies, drugs, bone marrow and kidney transplantation, pregnancy, and systemic diseases. The recently recognized disorders of complement regulation will be outlined in this review.
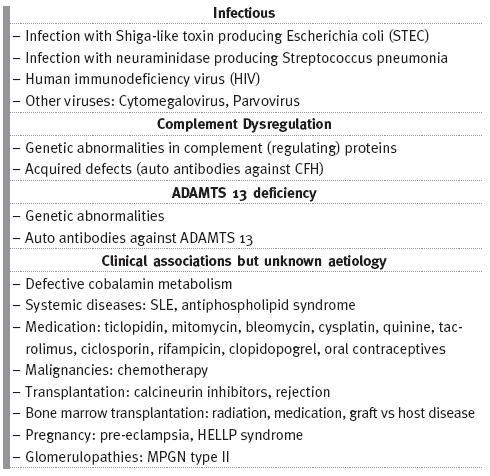
]]> The European Pediatric Research Study group for HUS proposed a new classification to TMA based on its cause (Table I)18,19 which distinguishes different TMAs, allowing guided investigation into its diagnosis and treatment.
Table I
Causes of thrombotic microangiopathy
aHUS: A DISEASE OF ALTERNATIVE COMPLEMENT PATHWAY DYSREGULATION
The human complement system is part of the innate immunity. Three activation pathways are recognised: the classical pathway, the mannose binding lectin pathway, and the alternative pathway. In aHUS, the alternative pathway is mostly affected. ]]>
Since 1974, reduced serum levels of complement fraction C3 with normal levels of C4 have been reported in patients with aHUS20-23 but it was only in the last decade that a clear link was demonstrated between aHUS and genetic abnormalities in complement (regulating) genes. The most frequently reported mutations (50-60%) are in the gene encoding complement factor H ( CFH), a plasma protein synthesized by the liver which downregulates alternative pathway activation24. More than 100 mutations have been described25. Many patients with heterozygous CFH mutations have normal CFH protein level but abnormal function.
Less frequently mutations in the genes encoding membrane cofactor protein (MCP) and complement factor I ( CFI) are present. MCP or CD46 is a widely expressed transmembrane glycoprotein that inhibits complement activation in host cells by serving as a membrane-bound cofactor for CFI to cleave C3b and C4b. About 35 different mutations have been described, mostly heterozygous. They are responsible for 10 to 15% of patients presenting with aHUS26,27.
CFI is a 2-chain serine predominantly synthesized by the liver that downregulates the alternative pathway by cleaving C3b, but it is efficient only in the presence of cofactor proteins (i.e. CFH and MCP).
Between 5% and 13% of aHUS patients have an CFI mutation, and approximately 25 different mutations have been reported, all heterozygous26,28-31.
More recently, a subgroup of aHUS patients without mutations in the genes encoding complement regulatory proteins but showing persistent activation of the alternative pathway with very low serum levels of C3 and normal or elevated serum levels of C4 have been identified. They were called gain-of-function mutations because they result in enhanced formation of C3bBb convertase or increased resistance to inactivation by complement regulatory proteins32. Mutations in complement factor B ( CFB ) and complement C3 ( C3) were proposed and are reported in 1 to 2% and in 4 to 10% respectively of aHUS patients, with heterozygous mutations, usually with low C3 levels 26,33.
There is a growing list of the mutations, polymorphisms and other complement abnormalities that alone or in combination account for about 10% of all aHUS, especially of CFI gene mutations with either CFH or MCP gene mutations. Mutations in the gene THBD encoding thrombomodulin, a membrane-bound glycoprotein with anticoagulant properties that modulates complement activation on cell surfaces, have been very also associated to aHUS34. These mutations may be acquired as autosomal recessive cases or autosomal dominant but the prognosis is not influenced by the inheritance11. The absence of a familiar history does not exclude the possibility of a genetic transmission because of an incomplete penetrance of the disease. The penetrance of disease among carriers of mutations in CFH, CFI, and MCP is approximately 50 to 60%35. This indicates that the genetic aberrations are probably important for the development of aHUS, but not the sole cause; an environmental factor, such as a complement trigger, is probably needed to develop the disease36.
Antibodies against factor H ( style='font-size:12.0pt;font-family:"Verdana","sans-serif"; mso-bidi-font-family:ArialMT;color:black;mso-ansi-language:PT;mso-bidi-language: AR-SA'>α FH) have also been observed in patients with aHUS, and in most cases in association with homozygous deletion of the genes encoding complement factor H-related proteins 1 and 3 ( CFHR1 and CFHR3)37.
]]> INVESTIGATION IN AHUS PATIENTSThe screening for all mutations mentioned above takes time and does not influence the initial treatment which should be started as soon as possible, but the analysis of the specific aHUS predisposing defects may help to establish differential diagnosis when the clinical presentation is ambiguous. It also influences long-term clinical management of affected patients, namely when considering transplantation38-41. STEC infection has to be ruled out because the classification of patients as STEC-associated or aHUS may be difficult: firstly, 10% of the patients do not present with diarrhoea42, and STEC-HUS can occur in adults as well, as in the German outbreak.
Such patients, if not tested for STEC, could erroneously be classified as aHUS. Secondly, STEC infection criteria (positive PCR for Stx genes in stools and/or circulating anti-lipopolysaccharides antibodies) are negative in approximately 15% of STEC-HUS patients43, leaving the physician concerned that the patient might in fact have aHUS. Thirdly, gastroenteritis was the triggering event in up to 28% of aHUS patients, including patients with
CFH, IF, or MCP mutations, in the French paediatric series44.
To select patients who should be analysed for complement mutations, the European Pediatric Research Study group for HUS suggests all patients who present with atypical features, regardless of whether they have typical signs such as a prodromal diarrhoeal illness41,45. Mutational screening should be performed in the complement genes that have been associated with aHUS (CFH, CFI, MCP, C3, CFB, and THBD), irrespective of serum C3, CFH, or CFI levels because their deficiency is not enough to exclude the diagnosis. It must be stressed that most assays measure the presence of the protein and not the activity. Moreover, abnormalities in complement regulation may only occur at the level of the endothelial cell surface, and not systemically. Therefore, serum levels may be normal in patients with complement dysregulation16,38. Moreover, in 40% of patients no mutation is found (un-genotyped patients).
Both aFH and ADAMTS13 should be searched for, the latter to exclude TTP. The possibilities of a rare cause of aHUS, such as HIV infection, pregnancy, or cobalamin deficiency, should be considered and investigated at presentation36.
An overview of the investigations to be performed in patients with aHUS is shown Table II.
Table II
]]> Investigations that should be performed in patients presenting with HUS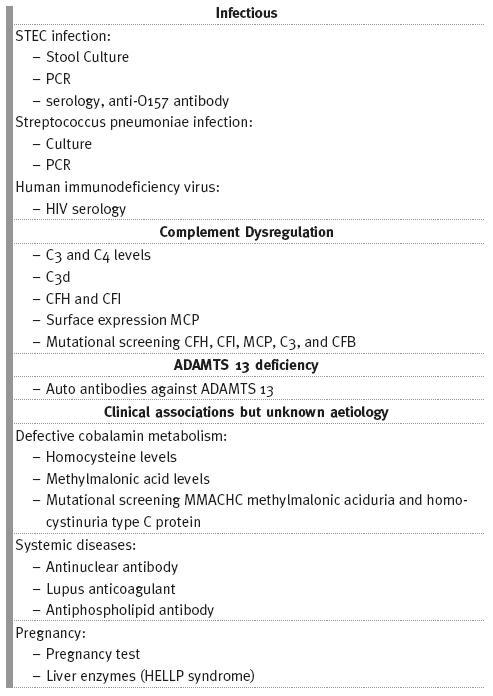
TREATMENT OPTIONS
Plasmatherapies
For a long time, the results of studies into therole of plasmapheresis in HUS have been controversial, because trials did not distinguish between STECHUS (where plasmapheresis seems not to change prognosis) from aHUS. Based on cohort studies that showed a 25 to 50% decrease in mortality rate since the introduction of plasma therapies36, guidelines propose the beginning of plasmapheresis within 24 hours of diagnosis41,45,46.
Exchange 1.5 times the expected plasma volume (60 to 75 ml/kg) and replace plasma with fresh frozen plasma or virus-inactivated pooled plasma is suggested. Plasmapheresis should be performed daily for five days, then five sessions a week for two weeks, and then three times a week for two weeks18. The total treatment time is not determinate, but recommendations state that treatment should be continued for at least two days after complete remission has been achieved. The dose and frequency may be reduced to weekly or biweekly intervals if plasma therapy appears to be successful. The parameters proposed to define patient remission are platelet count and lactate dehydrogenase levels in serum, since haptoglobin levels often remain decreased after achieving haematological remission. Some aHUS patients will remain plasma dependent and need chronic plasma treatment to stay in remission. It is also suggested that intercurrent infections and vaccinations can trigger a relapse of aHUS47,48 for which plasmapheresis should be started again or intensified.
For patients with isolated MCP mutation, plasma therapy has limited value since MCP is a membranebound protein, meaning the defect cannot be substituted by plasma therapy. In these cases, remission is achieved in 80 to 90% of these patients plasma therapy36. However, it is important to stress that by the time of first presentation, it is not known which complement genes are involved in the pathogenesis of aHUS and since it is recognised that combined mutations can occur, plasma therapy remains the first choice for treatment.
Plasmapheresis is the optimal treatment for patients with aFH, associated with steroids and immunosuppressive treatment (azathioprine, cyclophosphamide, mycophenolate mofetil, or rituximab) to prevent the redevelopment of antibodies after plasmapheresis cessation49,50.
If plasmapheresis is not available or cannot be applied immediately in the acute phase, plasma infusion should be started25. When plasma infusion is used instead of plasmapheresis, the suggested dosage is 30 to 40 ml/kg initially and 10 to 20 ml/kg per day thereafter16 because of the risks associated with the infusion of volume in patients who are already hypertensive and overloaded due to renal impairment.
]]> It can also be used after the initial period of plasmapheresis unless the tests have demonstrated that antibodies are the cause of the disease.Plasmapheresis also plays an important role in transplantation, as stated above.
In addition to plasmapheresis, avoiding triggers of endothelial injury, such as hypertension and hypercholesterolemia, by adequate blood pressure control and the use of statins are important treatment options in the acute phase of the disease and should be maintained once in remission51.
Transplantation
Kidney transplantation
Transplantation has also been controversial in aHUS because of the elevated risk of graft loss in the first year from 38%44 to 83%52 depending on the studies. This is attributed to an elevated risk of recurrence but also to a higher proportion of acute rejections53.
The underlying genetic defect predicts a different risk of recurrence. The biggest risk is seen in patients with a CFH mutation with a recurrence rate of 75 to 90%, followed by patients with a CFI with 45 to 80% and C3 mutation with 40 to 70%. Recurrences have been reported in patients with CFB and THBD mutations, as well54. Mutation in the gene encoding the membrane-bound MCP is at the lowest risk of developing a disease recurrence in the graft (<20%)36. As the graft has normal MCP levels, it would be expected that the risk would be zero. Nevertheless, cases have been reported26,55 probably because of the association of other mutations26.
A high proportion of vascular thrombosis has also been observed, probably because of the thrombogenic role of complement dysregulation26. The French paediatric series44 reported that of the 24 renal transplants performed in 15 aHUS children, 16 (67%) failed, and 66% of patients had at least one graft failure. Of the 16 graft failures, eight (50%) were due to graft vascular thrombosis 0-45 days after surgery.
]]> Prevention of post-transplant aHUS recurrenceIn the attempt to decrease recurrence of the disease, several hypotheses have been studied. Bilateral nephrectomy has been proposed because of a French cohort of non-genotyped adult patients who reported an increased risk of post-transplant recurrence in patients who still had their native kidneys compared with those who havent55. It was not observed in another series32: among genotyped aHUS patients, although 93% (14/15) of the patients in a French paediatric group44 had bilateral nephrectomy for hypertension before transplantation, the post-transplant recurrence rate was not lower than that reported in other genotyped cohorts38,51. Therefore, it is doubtful that pretransplant bilateral nephrectomy is beneficial in preventing post-transplant recurrence.
The avoidance of calcineurin inhibitors as also been proposed since de novo TMA have been reported with the use of these drugs however it was not associated with an increased incidence of HUS recurrence14,53,57.
Further, HUS recurrence has been reported in aHUS patients treated by sirolimus,
including one with MCP mutation54. In the consensus recommendation of 200946 no specific guidance is given about immunosuppression protocols, meaning aHUS was not considered per se a specific contraindication for treatment with calcineurin inhibitors.
Furthermore initial immunosuppression with mammalian target of rapamycin inhibitors is not encouraged in the absence of clear clinical advantages because of the possible association with impaired wound healing and delayed graft function58.
Finally, treatment with IVIG has occasionally been reported as efficient in aHUS patients, for instance in one child with non-genotyped aHUS59, or to treat post transplant recurrence in a child with CFI and CFB mutation (1 g/kgevery three weeks for five and a half years43 or in a patient with MCP mutation54.
Neutralisation of C5a complement activation product may be one of mechanism of action of IVIG60.
Living-related kidney transplantation
]]> aHUS has been considered a contraindication for living related kidney transplantation, not only for the high risk of aggressive relapse in graft61 but also because of the risk for the related donor who could himself develop HUS sometime after kidney donation.It was reported in four donors aged 21-31 years old, who donated a kidney to one of their children or siblings and had HUS three weeks to 10 months after donation62-64. It was also demonstrated in one patient who had HUS shortly after unilateral nephrectomy secondary to a traffic accident and was discovered to have a CFH mutation. The haemodynamic changes induced by unilateral nephrectomy could be the trigger for HUS in the donor with a predisposing genetic anomaly52. Even when no mutation is found, donation is not recommended because some polymorphisms have been linked to aHUS, as well as still unknown mutations.
Combined liver-kidney transplantation
The fact that the most frequent mutations occur in two circulating proteins synthesised by the liver, CFH and CFI, meant the advent of liver transplantation created great expectations. It would theoretically restore normal complement regulation and prevent disease recurrence. However, the first three combined liver-kidney transplantations in children with CFH deficiency occurred between 2002-2005 were disappointing, as the three died soon after. Autopsy of the liver revealed diffuse thrombotic and ischaemic lesions, most likely due to the thrombogenic effect of complement activation products deposited on the microvasculature of the liver after transplantation. Taking into account that liver transplantation might trigger intense local complement activation, it was suggested that liver transplantation should be performed under intensive pre-and perioperative plasma therapy to correct complement dysregulation. The first successful combined liver-kidney transplantation was done under such protocol and was reported in 200646.
This increased the bioavailability of functional CFH during the critical period needed for the liver graft to recover synthetic functions and, at the same time, removed the endogenous mutant CFH. In addition, posttransplant anticoagulation with low-molecularweight heparin at prophylactic dosages and low-dose aspirin was used in each of the successful procedures46.
This knowledge led a consensus group to propose protocols to make isolated kidney and combined-kidney transplantation46.
However, risks associated with this procedure still remain, and assessment of the risk/benefit ratio requires careful and individual attention. In addition, in the absence of a noted mutation, comprising a sizable fraction of patients with aHUS, liver-kidney transplantation should be avoided46.
Emerging new therapies
Concentrated CFH
]]> In patients with genetic CFH abnormalities it seems obvious to give normal CFH. A human plasma-derived CFH concentrate has been developed commercially with that intention, and it received the European orphan drug designation in January 2007. Substitution with CFH concentrate is a therapeutic option for patients with quantitative and functional CFH deficiency but it will have to be taken into account that such commercial concentrates have a short half-life.The same rationale applies to aHUS associated with CFI gene mutations but a CFI concentrate is still not available34.
Eculizumab
Eculizumab is a monoclonal antibody that inhibits the production of the terminal complement components C5a and the membrane attack complex C5b-9 by binding to complement protein C5. It has been approved since March 2007 for the treatment of patients with paroxysmal nocturnal haemoglobinuria (PNH). In September 2011 the U.S. Food and Drug Administration (FDA) also approved it for the treatment of aHUS patients based on its ability to prevent formation of C5a and the terminal complement complex inhibiting complement-mediated thrombotic microangiopathy.
By March 2012, 21 cases were reported in the literature of aHUS treated with eculizumab, including eight cases in recurrence in transplanted kidney, and three in prophylactic treatment after kidney transplant.
Eighteen patients went into complete or partial remission with functional kidney recovery and no need for subsequent renal replacement therapy. The longest period of remission has been observed in a patient treated with eculizumab for 28 months with no evidence of aHUS recurrence. Relapses after eculizumab treatment have only been seen when the treatment was discontinued or in patients who received a single dose65.
In addition to the above case reports, two prospective clinical trials have been conducted to evaluate the safety and efficacy of eculizumab use in aHUS66,67. In one study, seventeen patients with aHUS resistant to or intolerant of plasma therapy were treated with eculizumab for a minimum of 26 weeks. These patients showed decreased signs of TMA activity, including improvement in platelet counts and eGFR. In a second study, twenty patients with aHUS undergoing chronic plasma exchange or plasma infusion therapy were treated with eculizumab for a minimum of 26 weeks.
Adverse effects that were most frequently reported were hypertension, upper respiratory tract infection and diarrhoea. Meningococcal disease has been reported from PNH use and it was attributed to the impaired capacity for opsonization and clearance of encapsulated organisms68 Patients should be vaccinated at least two weeks before the start of the treatment. As vaccination does not protect against all serotypes, both patients and physicians should be aware of early signs of meningococcal infection69. Attention also has to be paid to patients treated with immunosuppressive drugs, as these therapies can reduce the response upon vaccination.
]]> In STEC-HUS patients, eculizumab is not indicated as a treatment option.Eculizumab is administered as an intravenous infusion. The recommended dosing for adult patients with aHUS is 900 mg weekly for the first four weeks, followed by 1200 mg weekly one week later, and 1200 mg every two weeks thereafter. The dosage regimen for paediatric patients is based upon body weight70.
Turning to the use of eculizumab in transplantation, a protocol has been proposed in pre-emptive use for isolated kidney transplantation71, which introduces some changes to previous protocols performed before eculizumab approval, but more studies are required to support the evidence of its use.
CONCLUSION
aHUS will always be a challenge to diagnose because it overlaps other TMAs. A differential diagnosis is crucial to the management of the entity. The knowledge of mutations associated to the regulators of alternative pathway was an important step in moving forward. Patients who should be studied have to be selected before starting treatment. Plasmapheresis is still the first choice but until now eculizumab has not been considered. This drug may represent a new hope for the treatment of primary disease and for recurrence after renal transplantation. Furthermore, although eculizumab is very expensive, it is expected to free dependent patients from plasmapheresis which is also an expensive treatment, with the advantage of granting a better quality of life.
References
1. Symmers WSC. Thrombotic microangiopathic haemolytic anemia (thrombotic microangiopathy). Br Med J 1952;2:542-544 [ Links ]
2. Furlan M, Robles R, Galbusera M, et al . Von Willebrand factor-cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic-uremic syndrome. N Engl J Med 1998;339:1578-1584 [ Links ]
3. Tsai HM, Lian ECY. Antibodies to von-Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998;339:1585-1594 [ Links ]
4. Levy GG, Nichols WC, Lian EC, et al . Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001;413:488-494 [ Links ]
5. Bianchi V, Robles R, Alberio L, et al . Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood 2002;100:710-71 [ Links ]
6. Dragon-Durey MA, Frémeaux-Bacchi V. Atypical haemolytic uraemic syndrome and mutations in complement regulator genes. Springer Semin Immunopathol 2005;27:359-374 [ Links ]
7. Caprioli J, Peng L, Remuzzi G. The hemolytic uremic syndromes. Curr Opin Crit Care 2005;11:487-492 [ Links ]
8. Mannucci PM, Franchini M. Advantages and limits of ADAMTS13 testing in the prognostic assessment of thrombotic thrombocytopenic purpura. Presse Med 2012;41:e157-62 [ Links ]
9. Frank C, Werber D, Cramer JP, et al . Epidemic profile of Shiga-toxinproducing Escherichia coli O104:H4 outbreak in Germany. N Engl J Med 2011;365:1771-80 [ Links ]
10. Michael M, Elliott EJ, Craig JC, et al . Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a systematic review of randomized controlled trials. Am J Kidney Dis 2009;53:259-72 [ Links ]
11. Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol 2005;16:1035-50 [ Links ]
12. Garg AX, Suri RS, Barrowman N, et al . Long-term renal prognosis of diarrhea associated hemolytic uremic syndrome: A systematic review, meta-analysis and metaregression. J Am Soc Nephrol 2003;290:1360-70 [ Links ]
13. Kemper MJ. Outbreak of hemolytic uremic syndrome caused by E. coli O104:H4 in Germany: a pediatric perspective. Pediatr Nephrol 2012;27:161-4 [ Links ]
14. Loirat C, Niaudet P. The risk of recurrence of hemolytic uremic syndrome after renal transplantation in children. Pediatr Nephrol 2003;18:1095-1101 [ Links ]
15. Terrell DR, Williams LA, Vesely SK, et al . The incidence of thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: all patients, idiopathic patients, and patients with severe ADAMTS-13 deficiency. J Thromb Haemost 2005;3:1432-6 [ Links ]
16. Kavanagh D, Goodship TH, Richards A. Atypical haemolytic uraemic syndrome. Br Med Bull 2006;77-78:5-22 [ Links ]
17. Jokiranta TS, Zipfel PF, Fremeaux-Bacchi V, et al . Where next with atypical hemolytic uremic syndrome? Mol Immunol 2007;44:3889-900 [ Links ]
18. Besbas N, Karpman D, Landau D, et al . A classification of hemolytic uremic syndrome and thrombotic thrombocytopenic purpura and related disorders. Kidney Int 2006; 70:423-31 [ Links ]
19. Keir L, Coward RJ. Advances in our understanding of the pathogenesis of glomerular thrombotic microangiopathy. Pediatr Nephrol 2011;26:523-33 [ Links ]
20. Cameron JS, Vick R. Letter: Plasma-C3 in haemolytic-uraemic syndrome and thrombotic thrombocytopenic purpura. Lancet 1973;2(7835):975 [ Links ]
21. Stühlinger W, Kourilsky O, Kanfer, Sraer JD. Hemolytic uremic syndrome: evidence forintravascular C3 activation. Lancet 1974;2:788-789
]]>22.
Carreras L, Romero R, requesens C, et al . Familial hypocomplementemic hemolytic uremic syndrome with HLA-A3,B7 haplotype. JAMA 1981;245:602-604 [ Links ]23. Noris M, Ruggenenti P, Perna A, et al. Hypocomplementemia discloses genetic predisposition to hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: a role of factor H abnormalities. J Am Soc Nephrol 1999;10:281-293 [ Links ]
24. Pichette V, Querin S, Schurch W, et al . Familial hemolytic-uremic syndrome and homozygous factor H deficiency. Am J kidney Dis 1994;24:936-941 [ Links ]
25. Saunders RE, Abarrategui-Garrido C, Fremeaux-Bacchi V, et al. The interactive Factor H-atypical haemolytic uremic syndrome mutation database and website: Update and integration of membrane cofactor protein and Factor I mutations with structural models. Hum Mutat 2007:28:222-234. Factor H aHUS mutation database 2007. Available at: http://www.fh-hus.org [ Links ]
26. Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transplantation 2008;12:619-629 [ Links ]
27. Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al . Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical haemolytic uremic syndrome.J Am Soc Nephrol 2006;17:2017-2025 [ Links ]
28. Kavanagh D, Richards A, Noris M, et al . Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol 2008;45:95-105
29. Nilsson SC, Karpman D, Vaziri-Sani F, et al . A mutation in factor I that is associated with atypical hemolytic uremic syndrome does not affect the function of factor I in complement regulation. Mol Immunol 2007;44:1835-1844 [ Links ]
30. Geelen J, van den Dries K, Roos A, et al . A missense mutation in factor I (IF) predisposes to atypical haemolytic uraemic syndrome. Pediatr Nephrol 2007;22:371-375 [ Links ]
31. Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, et al . Complement factor I: A susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet 2004;41:e84 [ Links ]
32.
33. Fremeaux-Bacchi V, Miller EC, Liszewski MK, et al . Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood 2008;112:4948-4952 [ Links ]
34. Delvaeye M, Noris M, De Vriese A, et al . Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med 2009;361:345-357 [ Links ]
35. Hirt-Minkowski P, Dickenmann M, Schifferli JB. Atypical Hemolytic Uremic Syndrome: Update on the Complement System and What Is New Nephron Clin Pract 2010;114:c219-c235 [ Links ]
36. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med 2009;361:1676-87 [ Links ]
37. Jozsi M, Licht C, Strobel S, et al . Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with FHR1/CFHR3 deficiency. Blood 2008;111:1512-1514 [ Links ]
38. Caprioli J, Noris M, Brioschi S, et al . Genetics of HUS: The impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006;108:1267-1279 [ Links ]
39. Kavanagh D, Goodship TH. Update on evaluating complement in hemolytic uremic syndrome. Curr Opin Nephrol Hypertens 2007;16:565-571 [ Links ]
40. Noris M, Remuzzi G. Genetic and biochemical testing hemolytic-uremic syndrome/thrombotic thrombocytopenic purpura. Clin J Am Soc Nephrol 2010;15:1844-1859 [ Links ]
41. Ariceta G, Besbas N, Johnson S, et al . Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol 2009;24:687-96 [ Links ]
42. Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005;365(9464):1073-86 [ Links ]
43. Lynn RM, O_Brien SJ, Taylor CM, et al . Childhood haemolytic uremic syndrome, United Kingdom and Ireland. Emerg Infect Dis 2005;11:590-596 [ Links ]
44. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al . Differential impact of complement mutations on clinical characteristics of atypical hemolytic uremic syndrome in children. J Am Soc Nephrol 2007;18:2392-2400 [ Links ]
45. Taylor CM, Machin S, Wigmore SJ, Goodship TH. Clinical practice guidelines for the management of atypical haemolytic uraemic syndrome in the United Kingdom. Br J Haematol 2010;148:37-47 [ Links ]
46. Saland JM, Ruggenenti P, Remuzzi G, Consensus Study Group: Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J Am Soc Nephrol 2008;20:940-949 [ Links ]
47. Geerdink LM, Westra D, van Wijk JAE, et al . Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol 2012;27:1283-91 [ Links ]
48. Olie KH, Goodship THJ, Verlaak R, et al . Posttransplantation cytomegalovirus-induced recurrence of atypical hemolytic uremic syndrome associated with a factor H mutation: Successful treatment with intensive plasma exchanges and ganciclovir. Am J Kidney Dis 2005;45:E12-E5 [ Links ]
49. Dragon-Durey MA, Loirat C, Cloarec S, et al . Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol 2005;16 555-563 [ Links ]
50. Jozsi M, Strobel S, Dahse HM, et al . Anti-factor H autoantibodies block C-terminal recognition unction of factor H in haemolytic uremic syndrome. Blood 2007;110:1516-1518 [ Links ]
51. Westra D, Wetzels JFM, Volokhina EB, et al . A new era in the diagnosis and treatment of atypical haemolytic uraemic syndrome. Neth J Med 2012;70:121-129 [ Links ]
52. Bresin E, Daina E, Noris M, et al . Outcome of renal transplantation in patients with non-Shiga-toxin associated hemolytic syndrome: Prognostic significance of genetic background. Clin J Am Soc Nephrol 2006;1:88-89 [ Links ]
53. Artz MA, Steenbergen EJ, Hoitsma AJ, et al . Renal transplantation in patients with hemolytic uremic syndrome: A high rate of recurrence and increased incidence of acute rejections. Transplantation 2003;76:821-826 [ Links ]
54. Loirat C, Fremeaux-Bacchi V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis 2011;6(1):60 [ Links ]
55. Fremeaux-Bacchi V, Arzouk N, Ferlicot S, et al . Recurrence of HUS due to CD46/MCP mutation after renal transplantation: A role for endothelial microchimerism. Am J Transplant 2007;7:2047-2051 [ Links ]
56. Lahlou A, Lang P, Charpentier B, et al. Hemolytic uremic syndrome. Recurrence after renal transplantation. Groupe Cooperatif de l_Ile-de-France (GCIF). Medicine (Baltimore) 2000;79:90-102 [ Links ]
57. Quan A, Sullivan EK, Alexander SR. Recurrence of hemolytic uremic syndrome after renal transplantation in children: A report of the North American Pediatric Renal Transplant Cooperative Study. Transplantation 2001;72:742-745 [ Links ]
58. Flechner SM, Kobashigawa J, Klintmalm G. Calcineurin inhibitor-sparing regimens in solid organ transplantation: focus on improving renal function and nephrotoxicity. Clin Transplant 2008;22:1-15 [ Links ]
59. Ito S, Okuyama K, Nakamura T, et al . Intravenous gamma globulin for thrombotic microangiopathy of unknown etiology. Pediatr Nephrol 2007;22:301-305 [ Links ]
60. Konrad S, Baumann U, Schmidt RE, Gessner JE. Intravenous immunoglobulin (IVIG) - mediated neutralisation of C5a: A direct mechanism of IVIG in the maintenance of a high Fc gammaRIIB to Fc gammaRIII expression ration on macrophages. Br J Haematol 2006;134:345-347 [ Links ]
61. Andrés A. Indications and contraindications for living kidney donations. Nefrologia 2010; 30(2):30-38 [ Links ]
62. Bergstein J, Michael A, Jr, Kellstrand C, Simmons R, Najarian J. Hemolytic-uremic syndrome in adult sisters. Transplantation 1974;17:487-490 [ Links ]
63. Kaplan BS, Papadimitriou M, Brezin JH, Tomlanovich SJ, Zulkharnain I. Renal transplantation in adults with autosomal recessive inheritance of haemolytic uremic syndrome. Am J Kidney Dis 1997;30:760-765 [ Links ]
64. Donne RL, Abbs I, Barany P, et al . Recurrence of haemolytic uremic syndrome after live related renal transplantation associated with subsequent de novo disease in the donor. Am J Kidney Dis 2002;40:E22 [ Links ]
65. Hodgkins K.S, Bobrowski A.E, Lane J.C, LangmanC.B. Clinical Grand Rounds: Atypical Hemolytic Uremic Syndrome. Am J Nephrol 2012;35:394-400 [ Links ]
66. Legendre CM, Babu S, Furman RR, et al. Safety and efficacy of eculizumab in aHUS patients resistant to plasma therapy: interim analysis from a phase II trial (SA-FC406) [abstract]. J Am Soc Nephrol 2010;21:93A [ Links ]
67. Muus P, Legendre C, Douglas K, et al. Safety and efficacy of eculizumab in aHUS patients on chronic plasma therapy: interim analysis of a phase II trial (F-PO1274)[abstract]. J Am Soc Nephrol 2010;21:402ª [ Links ]
68. Dmytrijuk A, Robie-Suh K, Cohen MH, et al. FDA report: eculizumab (Soliris) for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Oncologist 2008;13:993-1000 [ Links ]
69. Bouts A, Monnens L, Davin JC, et al. Insufficient protection by Neisseria meningitides vaccination alone during eculizumab therapy. Pediatr Nephrol 2011;26:1919-20 [ Links ]
70. http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125166s172lbl.pdf [ Links ]
71. Nester C, Stewart Z,Myers D, et al . Pre-emptive Eculizumab and Plasmapheresis for Renal Transplant in Atypical Hemolytic Uremic Syndrome Clin J Am Soc Nephrol 2011;6:1488-1494 [ Links ]
Dr Ana Farinha
Department of Nephrology
Centro Hospitalar de Setúbal, Portugal
Email: alpfarinha@yahoo.com.br
]]> Conflict of interest statement. None declared.
Received for publication: 27/08/2012
Accepted in revised form: 07/09/2012
]]>