Sandrina Martins1, Luís Ribeiro1, Miguel Fonte2, Rui Chorão1,3
1 S. Neurologia Pediátrica, HMPia, CHPorto;
2 S. Pediatria, CHTMADouro, Vila Real;
3 U. de Neurofisiologia Pediátrica, CHPorto
EEG case report
ABSTRACT
Introduction: Subacute sclerosing panencephalitis is a very rare disease in countries with measles vaccination programs, and is due to a persistent infection by a defective measles virus. The disease has a progressive fatal course.
]]> Case report: We describe the case of a 13 year-old boy with a progressive clinical picture of cognitive impairment, myoclonus, and pyramidal, extrapyramidal and cerebellar signs. The diagnosis was based upon clinical manifestations, the presence of characteristic periodic EEG discharges, and the demonstration of raised antibody titres against measles in the plasma and cerebrospinal fluid.Conclusions: Diagnosis of subacute sclerosing panencephalitis is based on clinical suspicion, very characteristic electroencephalographic abnormalities (typical periodic complexes) and raised antibodies to measles virus in the cerebrospinal fluid. It seldom occurs in patients with immunisation against measles, except in those with early onset of measles.
Keywords: Subacute sclerosing panencephalitis; electroencephalogram; periodic complexes; measles virus.
CASO CLÍNICO
Expõe-se o caso de um adolescente com 13 anos de idade na data em que foi referenciado ao nosso hospital.
Dos antecedentes patológicos, salientava-se história de sarampo aos sete meses de idade, com resolução do quadro sem intercorrências, e epilepsia focal idiopática com crises motoras do hemicorpo esquerdo de início aos 12 meses de idade e remissão aos três anos. Apresentava até à data do quadro clínico descrito um desenvolvimento psicomotor normal.
Aos 12 anos de idade começou a manifestar deterioração cognitiva, com perturbações mnésicas ligeiras e mau aproveitamento escolar. Posteriormente surgiram abalos com desequilíbrio e quedas bruscas. Foram ainda notadas posturas anómalas sustentadas. O quadro teve agravamento progressivo ao longo de um ano, particularmente célere nos três meses antes da admissão. Nesta altura apresentava abalos mais intensos e repetitivos, discurso incompreensível, incapacidade para a marcha sem apoio e perda de autonomia para as actividades de vida diária. O exame neurológico mostrava deterioração cognitiva; disartria marcada; síndrome piramidal irritativo bilateral, deficitário à esquerda; ataxia cerebelosa; distonia generalizada, mais marcada nos membros inferiores; e mioclonias faciais, distais dos membros superiores e axiais, estas muitas vezes com quedas associadas. O fundo ocular era normal.
Efectuou EEG que mostrou surtos generalizados de ondas lentas delta a 1,5-2 Hz, associando alguns potenciais abruptos (pontas), com duração até 2 segundos, que se repetiam periodicamente a intervalos de cerca de 5 segundos. (Figura 1). Por vezes apresentava mioclonias negativas concomitantes.
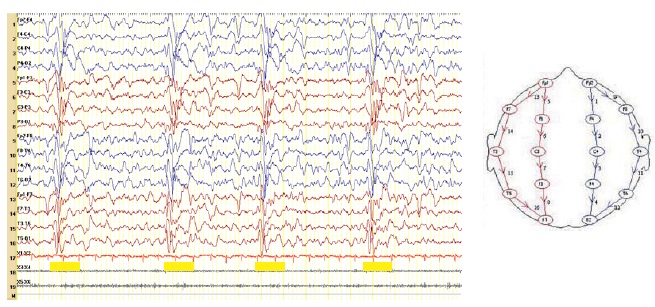 ]]>
Figura 1 - Complexos periódicos característicos da panencefalite esclerosante subaguda. (Base de tempo 15 mm/s)
]]>
Figura 1 - Complexos periódicos característicos da panencefalite esclerosante subaguda. (Base de tempo 15 mm/s)
Qual o seu diagnóstico?
DIAGNÓSTICO
Panencefalite Esclerosante Subaguda (pós-sarampo)
A ressonância magnética cerebral evidenciou ligeiro hipersinal em T2 da substância branca das regiões posteriores. A pesquisa de bandas oligoclonais no líquido cefalorraquidiano (LCR) foi positiva e foram detectados níveis elevados de anticorpos IgG anti-sarampo no LCR e soro. Estes resultados laboratoriais confirmaram, assim, o diagnóstico clínico e electroencefalográfico. Iniciou tratamento com isoprinosina oral, não tendo sido possível reunir condições para terapêutica intratecal. As mioclonias foram tratadas com associação de fármacos, que incluíram carbamazepina, clonazepam e levetiracetam; outros dos tratamentos sintomáticos incluíram tri-hexifenidilo, biperideno, baclofeno e toxina botulínica. Após um período de rápida deterioração clínica, houve estabilização dos défices neurológicos. Actualmente, com 17 anos de idade, apresenta défice cognitivo grave, quase sem linguagem, tetraparésia espástica com hemiplegia esquerda e distonia generalizada.
DISCUSSÃO
A Panencefalite Esclerosante Subaguda (PEES) é uma doença degenerativa do sistema nervoso central, lentamente progressiva, provocada pela infecção persistente por uma forma mutante do vírus do sarampo (1 -4).
]]> Era, já no passado, uma entidade pouco comum, tornando-se ainda mais rara nos países desenvolvidos desde a implementação da vacinação contra o sarampo(1 -4). É mais frequente no sexo masculino, havendo aumento da susceptibilidade quando a doença exantemática ocorre antes dos dois anos de idade(1,3 -5). O tempo de latência até ao aparecimento dos sintomas neurológicos varia habitualmente entre seis a 15 anos (3 -5).A forma de apresentação da doença é variável, embora classicamente se descreva uma evolução estereotipada (Estadios de Jabbour)(2). As alterações comportamentais e a deterioração cognitiva são frequentemente as primeiras manifestações da doença, a que se seguem, semanas ou meses depois, as mioclonias características. À medida que a doença progride associam-se sinais piramidais, cerebelosos e extrapiramidais, crises convulsivas, alterações visuais e, finalmente, um estado vegetativo que culmina com a morte do doente (1 -4).
O electroencefalograma (EEG) evidencia tipicamente complexos periódicos (a cada 4 a 15 segundos) de ondas delta de alta voltagem, com 0,5 a 2 segundos de duração(1-5), frequentemente concomitantes com espasmos mioclónicos axiais ou das extremidades. Este padrão do EEG pode, contudo, não ser evidente em fases precoces ou avançadas da doença(4). A ressonância magnética cerebral apresenta alterações inespecíficas, como hipersinal cortical ou subcortical na sequência T2 e atrofia cortical de grau variável(2,4,5). O LCR revela frequentemente hiperproteinorraquia, gamaglobulina elevada (com presença de bandas oligoclonais) e títulos elevados de anticorpos anti-sarampo (>1:4). Estes títulos são também elevados no soro (>1:256)(4). A biópsia cerebral, efectuada em casos excepcionais, mostra corpos de inclusão intranucleares ou citoplasmáticos que contêm antigénios virais(4,5).
À luz dos conhecimentos actuais, nenhum fármaco modifica a evolução da doença e, em 80% dos casos, a morte ocorre num período de três a cinco anos após o início dos sintomas, variando a apresentação clínica de quadros fulminantes a doença crónica prolongada(1). A terapêutica combinada com isoprinosina oral e interferão-alfa intratecal é a que tem demonstrado maiores benefícios(2,4,5). O tratamento sintomático visa o controlo das mioclonias, crises convulsivas, distonia e espasticidade(2,4). Considerando a evolução da doença e a ausência de tratamentos eficazes, a vacinação constitui a alternativa para evitar esta complicação tardia do sarampo, rara, mas invariavelmente fatal. Contudo, o seu risco mantém-se sobretudo nos casos de sarampo mais precoces.
O diagnóstico de PEES deve ser sempre considerado em crianças ou adolescentes com deterioração cognitiva, alterações de comportamento e mioclonias, sobretudo se houver história de sarampo em idades precoces.
BIBLIOGRAFIA
1. Garg RK. Subacute sclerosing panencephalitis. Post Grad Med J 2002; 78:63-70. [ Links ]
2. Campbell C, Levin S, Humphreys P, Walop W, Brannan R. Subacute sclerosing panencephalitis: Results of the Canadian Paediatric Surveillance Program and review of the literature. BMC Pediatrics 2005; 5:47.
3. Nunes ML, Costa JC, Stancher VM, Diament A, Arita F, Rosemberg S, Dyken P. Subacute sclerosing panencephalitis: Clinical aspects and prognosis. The Brazilian Registry. Arq Neuropsiquiatr 1999; 57(2A):176-81.
]]> 4. Bonthius JD, Stanek N, Grose C. Subacute sclerosing panencephalitis, a measles complication, in an internationally adopted child. Emerg Infect Disease 2000; 6:377-81.5. Tuncel D; Ozbek AE, Demirpolat G, Karabiber H. Subacute sclerosing panencephalitis with generalized seizures as a first symptom: a case report. Jpn J Infect Dis 2006; 59:317-9.
]]>