Anabela Bandeira1, Conceição Mota 2, Dulce Quelhas3, Marília Loureiro4, Esmeralda Martins1
1 S. Pediatria, H Maria Pia, CH Porto
2 S. Nefrologia Pediátrica, H Maria Pia, CH Porto
3 Unid. Biologia Clínica, CGM INSA
4 S. Cardiologia Pediátrica, H Maria Pia, CH Porto
]]> ABSTRACT
A 14 month-old boy presented with failure to thrive and severe mental and motor development delay. On physical examination he presented with severe axial hypotonia and dysmorphic syndrome: peculiar facies with small eyes, micrognathia, raised intermamilar distance. He also had multissistemic involvement with nephritic proteinuria, hypertrophy cardiomiopathy with pericardial effusion, raised transaminases, functional deficit of coagulation proteins and unspecific changes of retinal pigmentation. This case illustrates the typical presentation of congenital disorder of glycosilation (CDG) type Ia.
Keywords: congenital disorder of glycosilation, glycosylated transferring.
Criança, do sexo masculino, com 14 meses de idade, internado no primeiro ano de vida por má evolução ponderal e proteinúria nefrótica.
Trata-se de um primeiro filho de um casal jovem, não consanguíneo. Sem doenças heredofamiliares conhecidas.
Dos antecedentes fisiológicos: gravidez vigiada, sem intercorrências. Parto distócico por cesariana às 37 semanas de gestação, com índice de Apgar 6/8. Somatometria ao nascimento adequada à idade gestacional (peso 2610 g, P5; comprimento 47 cm, P10; perímetro cefálico 34,5 cm, P25). Leite adaptado desde o nascimento. Realizou rastreio endócrino-metabólico ao quinto dia de vida que foi normal.
Admitido na Unidade de Cuidados Intensivos (UCI) ao nascimento por gemido, necessidade de reanimação e edemas acentuados dos membros inferiores.
Ao exame físico: fácies particular com olhos pequenos, micrognatia, aumento da distância intermamilar e curvatura medial da falange distal do 5º dedo das mãos. Hipotonia axial marcada (Figura 1).
]]>
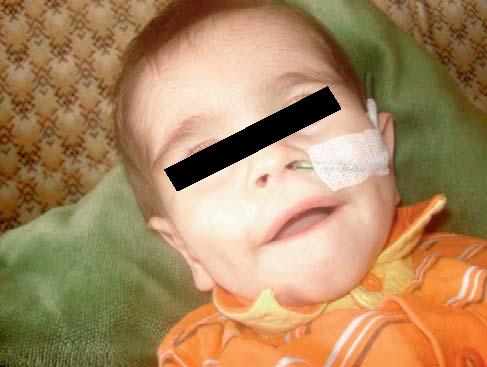
FIGURA 1
Teve alta da UCI com diagnóstico de síndrome dismorfico em estudo, sepsis clínica sem atingimento meníngeo. Completou 14 dias de antibiótico endovenoso (penicilina e gentamicina).
Do estudo realizado: cariotipo 46, XY; amónia, lactato e piruvato sem alterações; cromatografia dos aminoácidos sericos e urinários sem alterações, cromatografia dos ácidos orgânicos sem alterações, 7 dehidrocolesterol sem alterações, ácidos gordos de cadeia muito longa sem alterações, pesquisa de mutação para síndrome Prader Willi negativa.
Novo internamento aos 4 meses de vida por desnutrição grave, diarreia crónica e ITU por E.coli. Cumpriu 10 dias de cefuroxime e gentamicina. Ao exame físico foi constatada má evolução estaturo-ponderal com peso <<P5; comprimento <<P5 e perímetro cefálico <<P5; assim como atraso do desenvolvimento psico motor.
Analiticamente foi detectada proteinúria nefrótica com função renal conservada mas com atenuação da diferenciação parenquimosinusal bilateral e hiperecogenicidade parenquimatosa; cardiomiopatia hipertrofica com derrame pericárdio de moderado volume; elevação das transaminases, sem colestase e sem alteração da função de síntese hepática.
Qual o seu diagnóstico?
]]> COMENTÁRIOS
Perante um lactente com síndrome dismorfico e manifestações clínicas multissistémicas, podemos pensar em várias doenças genéticas. As malformações múltiplas podem ser decorrentes de cromossomopatias (6,1% dos casos), de mutações génicas (7,5% dos pacientes), de origem ambiental (6,5%) ou de origem multifactorial (20%). Perante a suspeita de doença hereditária do metabolismo, o atingimento sistémico obriga a pensar no diagnóstico de um defeito congénito da glicosilação (CDG) ou em citopatia mitocondrial.
Os CDG, anteriormente conhecidos como síndrome de glicoproteínas deficientes em carbohidratos, são um grupo de doenças autossómicas recessivas que afectam a síntese das glicoproteínas.(1) Estas patologias (com uma frequência estimada entre 1/50.000 e 1/100.000) são caracterizadas pelo envolvimento neurológico e/ou multi-orgânico.
Os síndromes CDG estão associados a diferentes deficiências enzimáticas, das quais a mais comum é a deficiência de fosfomanomutase (correspondendo ao CDG tipo Ia e que representa 70% dos casos).
O atraso psicomotor é o sinal clínico mais comum.(2) Outros sinais frequentemente associados de forma variável são: anomalias lipocutâneas (pele tipo casca de laranja, distribuição anormal da gordura), atrofia olivo-ponto-cerebelosa, anomalias ósseas, mamilos invertidos, anomalias da coagulação e citólise/fibrose hepática. Pode surgir estrabismo, proteinúria nefrótica, hipotonia e distúrbios cerebelares e também cardiomiopatia e derrame pericárdico.(3)
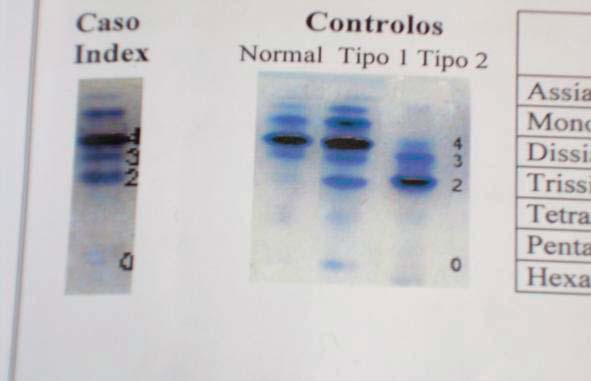
O rastreio bioquímico é baseado na demonstração de anomalias na glicosilação das glicoproteínas séricas através da focagem isoelétrica da transferrina (Figura 2). A determinação das actividades leucocitárias das enzimas responsáveis e a pesquisa de mutações nos genes correspondentes confirma o diagnóstico.
FIGURA 2
]]> Nesta criança o perfil de focagem isoeléctrica da transferrina foi compatível com CDG tipo 1. O diagnóstico foi confirmado pela presença de duas mutações em heterozigotia (p.R114H; p.D65Y) no gene PMM2, que codifica a enzima fosfomanomutase, CDG tipo 1 a. Actualmente com 14 meses de idade apresenta dismorfia já descrita, má evolução estaturo ponderal grave (peso e comprimento abaixo do percentil 5). Apresenta microcefalia com atraso do desenvolvimento psicomotor; cardiomiopatia hipertrófica com derrame pericardico de médio volume; proteinuria nefrotica; elevação das transaminases hepáticas e defeito funcional de proteínas da coagulação (anti trombina III e proteína C). Apresenta também alterações inespecíficas da pigmentação da retina, sem contacto ocular.
A ressonância magnética nuclear revelou atrofia cerebelar (Figura 3).
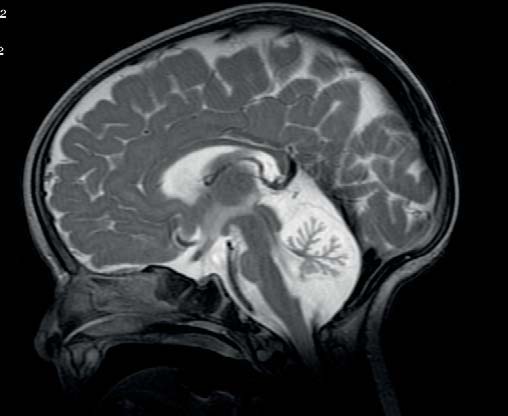
FIGURA 3
O tratamento farmacológico só é possível para CDG tipo 1b. Nesta criança, pela presença de diarreia crónica, foi inicialmente instituído tratamento com manose por se suspeitar de um CDG tipo 1 b. Após o diagnóstico molecular e falta de resposta clínica a manose foi retirada. O tratamento é essencialmente sintomático sendo importante a orientação dietética com aumento do aporte energético total.
Esta criança encontra-se medicada com furosemida; espironolactona; prednisolona e fenobarbital; assim como terapêutica anti refluxo. Faz suplementação calórica com MCT oil e necessita de sonda nasogástrica para alimentação. Faz fisioterapia duas vezes por semana.
As principais causas de mortalidade são as alterações hemorrágicas (trombose ou hemorragia) e as infecções. A cardiomiopatia hipertrofica pode determinar mau prognóstico.(4)
O diagnóstico pré-natal é possível sempre que seja identificada a mutação.
]]>BIBLIOGRAFIA
1. Aebi M, Helenius A, Schenk B, Barone R, Fiumara A, Berger EG, et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosilation: An updated nomenclature for CDG. Glycoconj J 1999; 16:669-71. [ Links ]
2. Barone R, Sturiale L, Fiumara A, Uziel G, Garozzo D, Jaeken J. Borderline mental development in a congenital disorder of glycosylation (CDG) type Ia patient with multisystemic involvement (intermediate phenotype). J Inherit Metab Dis 2007; 30:107. [ Links ]
3. Clayton PT, Winchester BG, Keir G. Hypertrophic obstructive cardiomyopathy in a neonate with the carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1992; 15:857-61. [ Links ]
4. Coman D, McGill J, MacDonald R, Morris D, Klingberg S, Jaeken J, et al. Congenital disorder of glycosylation type 1a: Three siblings with a mild neurological phenotype. J Clin Neurosci 2007;14:668-72. [ Links ]
]]>