Síndrome de Gorlin-Goltz: caso clínico
Gorlin-goltz syndrome: a case report
Sara DominguesI; Sandra PereiraI; Ângela MachadoI; Rui BezerraII; Fernando FigueiraII; Braga da CunhaII; Leonilde MachadoI
IS. Pediatria, U. Padre Américo, CH Tâmega e Sousa, 4564-007 Penafiel, Portugal. E-mail: spbaiao@gmail.com; anj.machado@gmail.com; bragadacunha@chts.min-saude.pt; 71612@chts.min-saude.pt
IIS. Estomatologia, U. Padre Américo, CH Tâmega e Sousa, 4564-007 Penafiel, Portugal. E-mail: ruibezerra@portugalmail.pt; 72449@chts.min-saude.pt
RESUMO
Introdução: O Síndrome de Gorlin-Goltz é uma condição hereditária autossómica dominante rara caracterizada por: car- cinomas basocelulares, queratoquistos odontogénicos, depres- sões palmo-plantares, calcificações da foice cerebral e malformações esqueléticas. Associa-se também a meduloblastoma e outras neoplasias.
Caso Clínico: Adolescente de 11 anos, com antecedentes de macrocefalia. Referenciada à consulta de estomatologia por quistos mandibulares recidivantes. O exame anátomo-patológico revelou tratarem-se de queratoquistos odontogénicos, pelo que é encaminhada para a consulta de pediatria. Objetivamente, apresentava macrocefalia e facies grosseiro com bossas frontais e hipertelorismo. Efetuou estudo genético, que detetou mutação do gene PTCH1, confirmando a suspeita clínica.
Discussão: Os queratoquistos odontogénicos são o achado mais representativo do síndrome de Gorlin-Goltz nas duas primeiras décadas de vida, sendo de extrema importância um elevado índice de suspeição por parte do pediatra, com especial relevância para as alterações minor. Após o estabelecimento do diagnóstico, é necessária uma equipa multidisciplinar para um adequado seguimento e tratamento atempado.
Palavras-chave: Carcinoma basocelular; macrocefalia; meduloblastoma; neoplasias cutâneas; quisto odontogénico; síndrome de Gorlin-Goltz; síndrome do nevo basocelular.
ABSTRACT
Introduction: The Gorlin-Goltz syndrome is a rare autosomal dominant inherited condition characterized by: basal cell carcinomas, odontogenic keratocysts, palmar or plantar pits, calcification of the falx cerebri and skeletal malformations. Medulloblastoma and other tumors are also associated with it.
]]> Case report: An 11-year-old female, with a history of macrocephaly, was referred to the Stomatology Department for recurrent mandibular cyst. Histopathological exam revealed odontogenic keratocysts, so the patient was referred to the Pediatric Department. Physical examination revealed macrocephaly and coarse face, with frontal bossing and hypertelorism. The genetic study performed detected a mutation in PTCH1 gene, confirming the clinical suspicion.Discussion: Odontogenic keratocysts are the most representative finding in Gorlin-Goltz Syndrome in the first two decades of life, therefore a high suspicion level is mandatory among pediatricians concerning detection of minor changes. After the diagnosis, a multidisciplinary team is required for adequate follow-up and timely treatment.
Key-words: Basal cell carcinoma; basal cell nevus syndrome; Gorlin syndrome; macrocephaly; medulloblastoma; multiple basal cell nevi; keratocysts; pits.
INTRODUÇÃO
O Síndrome de Gorlin-Goltz (SGG) é também conhecido como síndrome de Gorlin, síndrome dos basaliomas nevoides múltiplos, síndrome dos carcinomas basocelulares ou quinta facomatose(1). Foi descrito pela primeira vez em 1894 por W. Jarich e J.C. White, mas foi apenas em 1960 que R.J. Gorlin e R.W. Goltz estabeleceram a tríade clássica que o carateriza (carcinomas basocelulares múltiplos, queratoquistos mandibulares e costelas bífidas)(2,3). A prevalência na população geral, provavelmente subestimada, é de 1:57000 a 1:256000 e está descrito em todos os grupos étnicos, com distribuição semelhante no sexo masculino e feminino(1,3-7). É herdado de forma autossómica dominante, com elevada penetrância (aproximadamente 97%) e expressão fenotípica variável(1,4,7-9). A variabilidade de sinais e sintomas observada em indivíduos com a mesma mutação sugere que haja uma interação entre fatores genéticos e ambientais(3). Cerca de 30 a 50% dos doentes não tem história familiar, resultando de mutações de novo ou penetrância genética incompleta(3,5). Foram identificadas como responsáveis por este síndrome várias mutações do gene supressor tumoral PTCH1 (patched homolog 1 da Drosophila) localizado no braço longo do cromossoma nove (q22.3-q31)(3,5,9). Este gene codifica uma proteína transmembranar da via de sinalização Hedgehog, desempenhando um papel fundamental no controlo do crescimento e desenvolvimento dos tecidos normais(2,3).
Estão descritos mais de 100 sinais e sintomas relacionados com este síndrome(8).
As manifestações clínicas mais comuns são:
Carcinomas basocelulares múltiplos, presentes em 50-97% dos doentes, habitualmente a partir da adolescência e com maior incidência na raça branca e em áreas do corpo expostas a radiação solar ou a radioterapia(2,6);
]]> Queratoquistos odontogénicos, maioritariamente mandibulares (três quartos dos casos), estão presentes em 75-90% dos doentes, com pico de incidência na adolescência(6,8,10,11);Depressões palmares e/ou plantares, presentes em 50-70% dos doentes, são consideradas sinal patognomónico deste síndrome, mas raramente se desenvolvem antes da segunda década de vida(2,3,11);
Calcificações ectópicas da foice cerebral, presentes em mais de 90% dos doentes aos 20 anos de idade(10);
Malformações esqueléticas, como costelas bífidas (presen- tes em 38-60% dos doentes) e outros defeitos de fusão e modelagem das costelas; pectus excavatum, deformidade de Sprengel, sindactilia ou outros defeitos das mãos e pés e anomalias vertebrais (hemivértebras, fusão ou alongamento dos corpos vertebrais; casos de espinha bífida oculta também foram descritos)(2,5,8).
Aproximadamente 60% dos doentes apresentam dismorfias craniofaciais, como macrocefalia, bossas frontais, fenda lábio-palatina e hipertelorismo(9,10). O meduloblastoma ocorre em cerca de 5% das crianças com SGG, com uma predominância no sexo masculino (3:1)(2,8). Tem um pico de incidência aos dois anos de idade, ao contrário daquele que ocorre isoladamente (sete anos) e há evidência de que está associado a um melhor prognóstico(2,10). Quando o meduloblastoma se apresenta numa criança antes dos dois anos de idade, especialmente se for do subtipo desmoplásico é necessário investigar a presença do síndrome (12). Outras neoplasias estão associadas ao SGG, como fibromas cardíacos e ováricos (aproximadamente 2% e 20%, respetivamente); a ocorrência de linfomas e meningiomas também foi descrita(9,10).
O diagnóstico diferencial depende da apresentação inicial e inclui síndromes associados a hipertrofia (Síndrome de Sotos e de Beckwith-Wiedemann)(10) e alguns síndromes cutâneos raros como o síndrome de Bazex, tricoepitelioma múltiplo e síndrome de Muir-Torre(1,9).
É de extrema importância o reconhecimento atempado do SGG de forma a reduzir a morbimortalidade destes pacientes (principalmente devida às neoplasias cutâneas e cerebrais e à destruição e deformação oromaxilofacial)(9, 13).
CASO CLÍNICO
Adolescente de 11 anos, sexo feminino, segunda filha de um casal jovem, não consanguíneo, saudável. Fruto de gestação vigiada, sem intercorrências, de termo. Parto eutócico, com peso ao nascer 3474g (P50), comprimento 52cm (P90) e perímetro cefálico 36cm (P50-90). Seguida em consulta de pediatria geral na infância por macrocefalia. Evolução estaturo-ponderal no percentil 95. Desenvolvimento psico-motor adequado à idade.
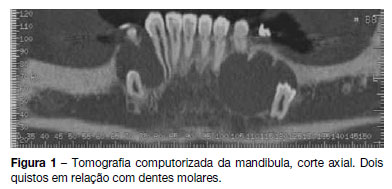
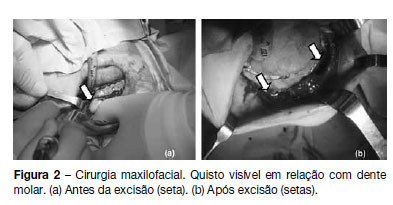
]]> Referenciada à consulta de estomatologia do nosso hospital em 2011 por quistos mandibulares recidivantes (já submetida a cirurgia excisional em 2008). Realizou tomografia computorizada da mandibula, que revelou a presença de dois quistos em relação com dentes molares (Figura 1) pelo que se procedeu a cirurgia excisional (Figura 2). O exame anátomo-patológico revelou tratarem-se de queratocistos odontogénicos, tendo sido encaminhada para a consulta de pediatria geral. Ao exame objetivo apresentava macrocefalia e peso e estatura acima do percentil 97, facies grosseiro, com bossas frontais, hipertelorismo, base do nariz alargada, prognatismo e mal oclusão dentária (Figura 3). Sem lesões da pele, nomeadamente, depressões palmares ou plantares, hiperqueratose ou lesões sugestivas de carcinoma de células basais; sem escoliose ou outras malformações esqueléticas aparentes. Efetuou estudo genético, que detetou uma mutação em heterozigotia no gene PTCH1 que não tinha ainda sido descrita (mutação c.2179dupT/p.Cys727LeufsX11) no exão 14, confirmando a suspeita clínica. Efetuou ecografia abdomino-pélvica, eletrocardiograma e ecocardiograma, que não apresentaram alterações. A ressonância magnética cerebral revelou discreta hipogenesia do corpo caloso.
]]>

A doente foi orientada para consulta de genética; a avaliação clinica e imagiológica (ortopantomografia) dos familiares em primeiro grau (pais e irmã de 18 anos), não revelou sinais sugestivos do síndrome. Aguarda observação por dermatologia e oftalmologia. Quando atingir a maioridade ou desejar engravidar, a doente deve ser encaminhada para consulta de Aconselhamento Genético.
DISCUSSÃO
O reconhecimento do SGG é um desafio, especialmente na criança, pois a maioria das alterações major não estão presentes antes da segunda-terceira década de vida (5, 12). No caso clínico apresentado, a adolescente tinha já sido seguida em consulta de pediatria para estudo da sua macrocefalia, mas na altura não apresentava ainda nenhum sinal clinico major sugestivo do síndrome. Estes sinais habitualmente desenvolvem-se ao longo do tempo(13), como foi o caso do aparecimento dos queratoquistos odontogénicos, anos mais tarde.
Em 1997, Kimonis(14) propôs uma modificação aos critérios antes definidos por Evans(15) para o diagnóstico do SGG, baseado em critérios major e minor. Para o diagnóstico, basta a presença de dois critérios major ou um major e dois minor(14) (ver Quadro I). A doente descrita apresenta um sinal major (queratoquistos odontogénicos) e dois minor (macrocefalia e malformações congénitas facies grosseiro, com bossas frontais e hipertelorismo), cumprindo os critérios de Kimonis.
]]>

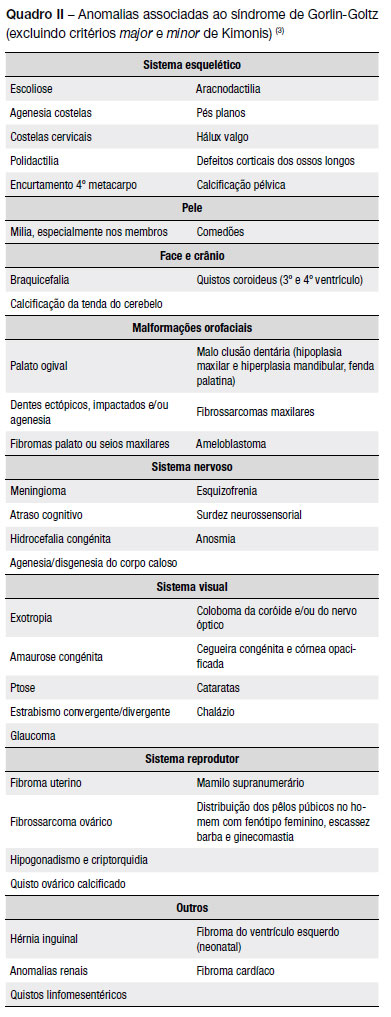
No Quadro II estão descritas outras anomalias associadas ao síndrome(3).
A demonstração da mutação no exão 14 do gene PTCH1 confirmou o diagnóstico (1). Dada a ausência de história familiar compatível, o exame físico e ortopantomografias dos familiares em primeiro grau não terem alterações de relevo, presume-se que tenha ocorrido uma mutação de novo.
Tendo em conta que estes pacientes são altamente sensíveis à radiação ionizante (5), o seu uso no caso apresentado foi minimizado, tendo-se realizado apenas exames complementares de diagnóstico inócuos para a adolescente (ultra-som e ressonância magnética). O ecocardiograma e a ecografia abdomino-pélvica permitiram a exclusão de fibromas cardíacos ou ováricos, malformações renais ou outras. A ressonância magnética permitiu a exclusão de tumores intracranianos e detetou uma hipogenesia do corpo caloso, aspecto associado ao síndrome(3). Dado ser um síndrome com atingimento de vários órgãos e sistemas, após o estabelecimento do diagnóstico, é necessária uma equipa multidisciplinar para um adequado seguimento e tratamento atempado(9). A paciente deverá manter seguimento regular por dermatologia (pelo menos anualmente, havendo mesmo quem recomende observação médica a cada 3-4 meses)(10). A ortopantomografia está indicada a cada 12-18 meses, a partir dos oito anos de idade(10).
Excisão cirúrgica, crioterapia, ablação com laser, terapia fotodinâmica e quimioterapia tópica (ácido delta-aminolevulínico, 5-fluorouracil, imiquimod a 5%) são opções terapêuticas nos carcinomas basocelulares; a radioterapia deve ser evitada (9,10). O uso tópico de antagonistas da proteína sonic hedgehog é promissor (10). Os análogos da vitamina A (retinóides) podem ser utilizados na quimioprevenção de lesões cutâneas novas, mas apresentam toxicidade importante(7-9).
]]> O tratamento dos queratoquistos é cirúrgico, devendo ser tratados de forma conservadora dada a sua agressividade e alta tendência a recorrência (30-60%)(5,7,8). O meduloblastoma necessita tratamento intensivo; os melhores resultados obtêm-se com recessão agressiva, quimio e radioterapia(1). Mas o risco relativo de neoplasias secundárias induzidas pela radioterapia (carcinomas basocelulares múltiplos, tumores intracranianos ou nasosinusais) é 20-39 vezes superior à da população geral, pelo que deve ser um tratamento preferencialmente de segunda linha, quando não há resposta à quimioterapia ou há recorrência do tumor(1,12). Estes doentes necessitam ainda de proteção solar estrita: devem utilizar óculos de sol com filtros de proteção 100% para raios ultravioleta, evitar exposição solar no horário mais nocivo, utilizar fator de proteção mínimo de 30 e repetir a aplicação a cada 2-3 horas e após o banho ou transpiração(1).O rastreio dos familiares e o aconselhamento genético são peremptórios(2). Apesar de cerca de 70 a 80% dos doentes terem um progenitor afetado, este pode não ser reconhecido, devido à enorme variabilidade fenotípica do síndrome(10); o primeiro individuo afetado na família pode ter apenas pequenas alterações do SGG, explicado pelo mosaicismo somático(13). O risco de transmissão para os descendentes é de 50% (1). O diagnóstico pré-natal é possível através da análise do ADN de células fetais obtidas por amniocentese ou biopsia de vilosidades coriónicas e pode ser útil na prevenção de complicações (prever a necessidade de parto distócico por macrocefalia e deteção de malformações congénitas graves ou fibromas cardíacos)(1).
A sobrevida destes doentes, exceto nos casos em que há meduloblastoma, é boa(5), sendo a maior preocupação o efeito estético do tratamento dos múltiplos carcinomas basocelulares e queratoquistos, com implicações quer a nível psicológico quer social(10).
CONCLUSÃO
Os queratocistos odontogénicos são o achado mais representativo do Síndrome de Gorlin-Goltz na primeira e segunda décadas de vida, pelo que é maioritariamente reconhecido entre dentistas e estomatologistas(2,9). Outros critérios major, como carcinomas basocelulares e depressões palmoplantares habitualmente ocorrem em idades mais avançadas(2). Assim, é de extrema importância um elevado índice de suspeição por parte do pediatra para o diagnóstico deste síndrome, dando especial relevância às alterações minor. No caso apresentado, a doente tinha sido já estudada por macrocefalia; apresentava ainda outros sinais minor (facies grosseiro com bossas frontais e hiper-telorismo)(14) mas a suspeita diagnóstica apenas surgiu aquando do aparecimento de queratoquistos odontogénicos recidivantes.
Após o estabelecimento do diagnóstico, é necessária uma equi- pa multidisciplinar para um adequado seguimento e tratamento atempado(9). O rastreio dos familiares e o aconselhamento genético são peremptórios(2). Estes doentes são extremamente sensíveis a radiação ionizante, com propensão ao desenvolvimento de basaliomas e meningiomas pelo que a exposição solar, a radiação X e a radioterapia devem ser evitadas(2).
REFERÊNCIAS BIBLIOGRÁFICAS
1. Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J Rare Dis 2008; 3:32-47. [ Links ]
2. Safronova MM, Arantes M, Lima I, Domingues S, Almeida M, Moniz P. Síndrome de Gorlin-Goltz Revisão das Características Neurorradiológicas e Maxilofaciais Ilustradas Com Dois Casos. Acta Med Port 2010; 23:1119-26. [ Links ]
3. Ortega-García-de Amezaga A, García-Arregui O, Zepeda-Nuño S, Acha-Sagredo A, Aguirre-Urizar JM. Gorlin-Goltz syndrome: Clinicopathologic aspects. Med Oral Patol Oral Cir Bucal 2008; 13:E338-43. [ Links ]
4. Berg D. Nevoid basal cell carcinoma syndrome. [Online] 2011 Sep 14, [Acedido em: 2012 Dec 12] Disponível em: URL: http://emedicine.medscape.com/article/1101146-overview. [ Links ]
5. Medeiros L, Ferreira JC. Síndrome de Gorlin-Goltz: revisão bibliográfica a propósito de um caso clínico. Rev Port Estomatol Cir Maxilofac 2006; 47:25-32. [ Links ]
6. Ljubenovic M, Ljubenovic D, Binic I, Jovanovic D, Stanojevic M. Gorlin-Goltz syndrome. Acta Dermatovenerol Alp Panonica Adriat 2007; 16:166-9. [ Links ]
7. Rosón-Gómez S, González-García R, Naval-Gías L, Sastre-Pérez J, Muñoz-Guerra MF, Díaz-González FJ. Síndrome de Gorlin-Goltz: Serie de 7 casos. Rev Esp Cir Oral Maxilofac 2009; 31:309-15. [ Links ]
8. Barankin B, Goldenberg G, Stern RS, Robinson JK. Nevoid basal cell carcinoma syndrome [Online] 2012. Oct 24, [Acedi- do em: 2012 Dec 15]. Disponível em: URL: http://www.uptodate.com/contents/nevoid-basal-cell-carcinomasyndrome/ [ Links ]
9. Kalogeropoulou C, Zampakis P, Kazantzi S, Kraniotis P, Mastronikolis NS. Gorlin-Goltz syndrome: incidental finding on routine ct scan following car accident. Cases J 2009; 2:9087. [ Links ]
10. Evans DG, Farndon PA. Nevoid Basal Cell Carcinoma Syndrome. 2002 Jun 20 [Updated 2011 Nov 3]. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1151/ [ Links ]
11. Díaz-Fernández JM, Infante-Cossío P, Belmonte-Caro R, Ruiz-Laza L, García-Perla AG, Gutiérrez-Pérez JL. Síndrome névico basocelular. Presentación de seis casos y revisión de la literatura. Med Oral Patol Oral Cir Bucal 2005; 10: E57-66. [ Links ]
12. Choudry Q, Patel HC, Gurusinghe NT, Evans DG. Radiation induced brain tumours in nevoid basal cell carcinoma syndrome: implications for treatment and surveillance. Childs Nerv Syst 2007; 23:133-6. [ Links ]
13. Veenstra-Knol HE, Scheewe JH, van der Vlist GJ, van Doorn ME, Ausems MG. Early recognition of basal cell naevus syndrome. Eur J Pediatr 2005; 164:126-30. [ Links ]
14. Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet 1997; 69:299-308. [ Links ]
15. Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet 1993; 30:460-4. [ Links ]
Endereço para correspondência ]]>
Sara DominguesRecebido a 09.06.2013 | Aceite a 30.08.2013
]]>