Caso dermatológico
Dermatology case
Fernando MotaI; Susana MachadoI,II,III; Manuela SeloresI,II,III,IV
I Department of Dermatology, Centro Hospitalar do Porto. 4099-001 Porto, Portugal. fernandojrmota@gmail.com; susanamlmachado@gmail.com; mselores@gmail.com
II Dermatology Research Unit, Centro Hospitalar do Porto. 4099-001 Porto, Portugal. susanamlmachado@gmail.com; mselores@gmail.com ]]>
III Instituto de Ciências Abel Salazar, Universidade do Porto. 4050-313 Porto, Portugal. susanamlmachado@gmail.com; mselores@gmail.com
IV Unit for Multidisciplinary Research in Biomedicine, Universidade do Porto. 4050-313 Porto, Portugal. mselores@gmail.com
RESUMO
A esclerose tuberosa é uma síndrome neurocutânea, hereditária, autossómica dominante, caracterizada por manifestações pleomórficas envolvendo m últiplos órgãos, incluindo a pele. O diagn óstico de esclerose tuberosa é clínico. As lesões cutâneas mais comuns nestes doentes s ão: máculas hipopigmentadas, habitualmente com forma elíptica, os angiofibromas, tipicamente envolvendo a região malar e placas de shagreen, mais frequentemente localizadas à região inferior do tronco.
No caso descrito, a doente foi referenciada à consulta de dermatologia por uma lesão atípica de localização incomum, que demonstra a importância da observação de toda a pele nesta patologia.
Palavras-chave: Esclerose tuberosa; placa de Shagreen; maculas hipopigmentadas; angiofibromas.
Tuberous sclerosis is an inherited neurocutaneous disorder characterized by pleomorphic features involving many organ systems, including the skin. The diagnosis of tuberous sclerosis is clinical. The most common cutaneous lesions are hypopigmented macules, also known as ash-leaf spots, which are usually elliptic in shape, angiofibromas, which typically involve the malar regions of the face, and shagreen patches, most commonly present over the lower trunk.
]]> In the present case, the patient was referred to the dermatology department due to an atypical lesion with an uncommon location, revealing the importance of a whole body examination in patients with this disease.Keywords: Tuberous sclerosis; Shagreen patch; hypopigmented macules; angiofibromas
Criança de dois anos, sexo feminino, com antecedentes de epilepsia no contexto de esclerose tuberosa.
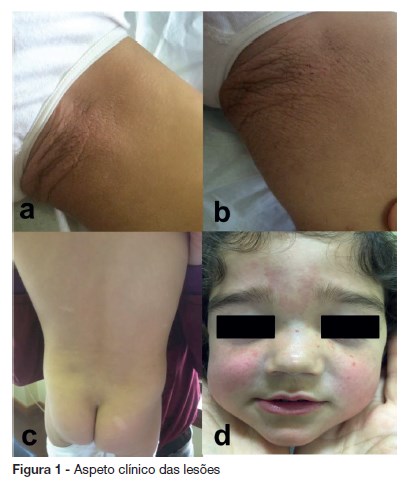
Foi referenciada à consulta de dermatologia pediátrica por uma placa na coxa esquerda desde os quatro meses de idade. Ao exame objetivo observava-se uma placa de colora ção idêntica à pele circundante, com 5 x 3,5 cm, de superfície rugosa e apergaminhada, na face interna da coxa esquerda (figura 1a e 1b).
]]> No restante exame físico identificaram-se máculas hipopigmentadas de cerca de dois cent ímetros de maior eixo, no abd ómen, dorso e nádegas (figura 1c) e algumas p ápulas eritematosas na face (figura 1d).
Qual o seu diagnóstico?
DIAGNÓSTICO
Placa de Shagreen (Esclerose Tuberosa).
A biópsia cutânea confirmou o diagnóstico clínico.
DISCUSSÃO
A esclerose tuberosa (ET) é uma síndrome neurocutânea, hereditária, autossómica dominante.1 Atinge ambos os sexos e todos os grupos étnicos.1 A incidência estimada é de um em cada 5000-10000 indivíduos.2
A doença é causada pela muta ção no gene TSC1 ou TSC2. Muta ções de novo são responsáveis por aproximadamente 80% dos casos de ET, sendo a mutação no gene TSC2 cerca de quatro vezes mais frequente nos casos esporádicos, enquanto nos casos familiares a prevalência é igual para ambos os genes.3 A ET tem uma expressão clínica muito vari ável, nomeadamente quanto à idade de aparecimento, gravidade da doen ça e diferentes sinais e sintomas que resultam de um gen ótipo espec ífico. Caracteriza-se pelo desenvolvimento de múltiplos tumores benignos hamartomas em vários órgãos, incluindo o cérebro, coração, pele, olho, rim, pulm ão e fígado, existindo um risco de malignidade superior nestes doentes.2
]]> Um grande número de doentes com ET têm epilepsia, e mais de 50% pode revelar défices cognitivos e dificuldades de aprendizagem. Estas altera ções neurológicas estão normalmente associadas a lesões cerebrais, incluindo hamartomas glioneuronais periventriculares, tuberomas corticais, astrocitomas subependimários de células gigantes e linhas de migração neuronal da substância branca detetadas em exames de imagem cerebral, nomeadamente a ressonância magnética.1,4A grande maioria dos doentes com ET (81 95%) tem uma ou mais lesões cutâneas características desta doença: maculas hipopigmentadas, que habitualmente têm uma forma el íptica; angiofibromas, tipicamente envolvendo a região malar bilateralmente; placas de shagreen, localizadas frequentemente à metade inferior do tronco.5,6 As lesões cutâneas n ão apresentam risco aumentado de maligniza ção, sendo que o seu número aumenta ao longo da puberdade e estabiliza na idade adulta. Neste caso, a lesão pela qual a doente foi referenciada tinha morfologia atípica e uma localização incomum para placa de shagreen, o que revela a import ância da observação da totalidade da pele para visualiza ção das restantes lesões cutâneas que permitiram estabelecer o diagnóstico, confirmado pela análise histológica.
O diagnóstico de ET baseia-se em critérios clínicos e/ou testes gen éticos. Os critérios clínicos foram publicados por Roach e revistos pela Tuberous Sclerosis Alliance e pelo National Institutes of Health em 1998. 7 O diagnóstico definitivo de ET é feito na presença de dois critérios major ou um major e dois minor. O diagnóstico é provável na presença de um critério major e um minor. Considera-se ET possível na presença de um critério major ou dois ou mais critérios minor. Os testes genéticos não são imprescindíveis para o diagnóstico se o doente cumprir os critérios clínicos; no entanto são úteis para estudo familiar ou em casos de ET prov ável ou possível devido à penetrância incompleta dependente da idade e a possibilidade de mosaicismo som ático.
Torna-se assim imprescindível para os clínicos conhecerem o espectro de manifesta ções clínicas inerentes a esta doen ça.
O tratamento das alterações dermatológicas desta doença, como os angiofibromas ou placas de shagreen, pode ser realizado com laser ou dermoabrasão.1,8 Estudos recentes com o uso tópico da rapamicina têm mostrado resultados promissores no tratamento de angiofibromas. 9
Estes doentes devem ser seguidos por equipas médicas multidisciplinares que deverão incluir neurologia, dermatologia, nefrologia, cardiologia, oftalmologia, genética médica e pneumologia.1,7,8
REFERÊNCIAS BIBLIOGRÁFICAS
1. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345. [ Links ]
]]>2. Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet 2008;372:657. [ Links ]
3. Au KS, Williams AT, Roach ES, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 2007;9:88. [ Links ]
4. Lyczkowski DA, Conant KD, Pulsifer MB, et al. Intrafamilial phenotypic variability in tuberous sclerosis complex. J Child Neurol 2007;22:1348. [ Links ]
5. Yates JR, Maclean C, Higgins JN, et al. The Tuberous Sclerosis 2000 Study: presentation, initial assessments and implications for diagnosis and management. Arch Dis Child 2011;96:1020. [ Links ]
6. Aldrich CS, Hong CH, Groves L, et al. Acral lesions in tuberous sclerosis complex: insights into pathogenesis. J Am Acad Dermatol 2010;63:244. [ Links ]
]]>7. Roach ESm Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998;13:624-8. [ Links ]
8. Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care 2007;21:108-14. [ Links ]
9. Balestri R, Neri I, Patrizi A, Angileri L, Ricci L, Magnano M. Analysis of current data on the use of topical rapamycin in the treatment of facial angiofibromas in tuberous sclerosis complex. J Eur Acad Dermatol Venereol 2015;29:14-20. [ Links ]
CORRESPONDENCE TO
Fernando Mota
Department of Dermatology ]]>
Centro Hospitalar do Porto
Largo Prof. Abel Salazar
4099-001 Porto
Email: fernandojrmota@gmail.com
Received for publication: 29.07.2016 Accepted in revised form: 04.10.2016
]]>