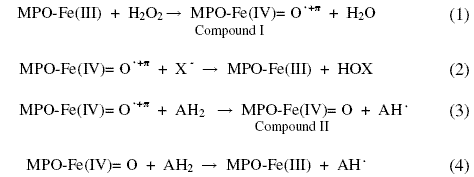
Yahya R. Tahboub,1,* Husam M. Abu-Soud2
1 Department of Applied Chemistry, Jordan University of Science and Technology, Irbid 22110, Jordan
2 Department of Obstetrics and Gynecology, The CS Mott Center for Human Growth and Development, Wayne State University, School of Medicine, Detroit, MI, USA
Abstract
Myeloperoxidase (MPO) is a neutrophil enzyme that employs hydrogen peroxide (H2O2) to catalyze the oxidation of halides and thiocyanate to their respective hypohalous acids. In this study, the inhibitory effect of nitrite (NO2-) on MPO-catalytic activity was investigated electrochemically. H2O2 consumption during steady-state catalysis was monitored amperometrically by a carbon fiber based H2O2-biosensor at 25 oC. Optimized initial concentrations were 50 nM MPO, 10 μM H2O2, and a selected halide or thiocyanate concentration from physiological range. Under these conditions, reactions were monophasic and rapid (complete H2O2 consumption occurs in < 10 s). As concentration of NO2- increases, reactions change to biphasic (rapid step followed by a slow step) and both steps have been inhibited by NO2-. Our results confirmed the inhibitory effect of NO2- and demonstrated for the first time that NO2- is a strong inhibitor towards MPO-catalyzed oxidation of iodide and bromide; and a weak inhibitor towards MPO-catalyzed oxidation of chloride and thiocyanate.
Keywords: nitric oxide, nitrite, myeloperoxidase, catalytic activity, H2O2-biosensor.
]]> Introduction
Nitric oxide (nitrogen monoxide, NO) is ever-present signaling molecule involved in the regulation of many processes, including activities of the cardiovascular, nervous and immune systems [1-4]. Stimulation of NO synthases during inflammatory processes represents a defense mechanism against invading organisms, although excessive formation of NO has been implicated in host tissue injury [5,6]. Nitrite (NO2-) is observed to accumulate upon nitric oxide synthase activation in many inflammatory diseases [7] and NO2- concentrations can be higher in tissues than those measured in plasma [8,9]. In healthy human subjects, NO2- can be detected at levels 0.5-3.6 μM in plasma [10,11], ~ 15 μM in respiratory tract lining fluids [12], 30-210 μM in saliva and 0.40-60 μM in gastric juice [13]. Extra cellular NO2- levels markedly increase during inflammatory processes reflecting increased NO production. For instance, a serum level of 36 μM has been reported in human immunodeficiency virus-infected patients with interstitial pneumonia [8].
Myeloperoxidase (MPO), a member of mammalian peroxidases, displays a crucial difference (within a wide range of biological processes) in its unique ability in catalyzing the H2O2-dependent peroxidation of halides and pseudo halides to produce antimicrobial agents and hypohalous acids [14-19].
The simplified mechanism that governs the catalytic activity of MPO can be represented by the classic peroxidases catalytic cycle, which is represented by Equations 1-4,
H2O2 reacts rapidly and reversibly with ground state (MPO-Fe(III)) and generates a ferryl π cation radical (MPO-Fe(IV)= O ·+π ) intermediate compound I [20,21]. Compound I is capable of oxidizing either halides and pseudo halides (X-) through a 2e- transition generating the ground state and the corresponding hypohalous acid (HOX). During turnover, compound I is also converted to peroxidase intermediate compound II (MPO-Fe(IV)=O) and MPO-Fe(III) ,respectively, because of the presence of exogenous (AH2) or endogenous electron donors [20-23]. Compound II is inactive in 2e oxidation of X- and is a longer lived intermediate whose decay to ground state is considered to be the rate-limiting step during steady-state catalysis [20,21]. Acceleration in compound II formation and decay has been noted with a series of organic and inorganic substrates [22-25].
The effect of NO2- on catalytic activity of MPO was reported by a number of research groups [26-32]. It has been demonstrated that MPO and other peroxidases can oxidize NO2- to a species capable of nitrating tyrosine and tyrosyl residues in protein [26-30]. Also, it was reported that NO2- is a poor substrate for ground state MPO and an inhibitor for its chlorination activity, and it is oxidized by two one-electron steps in the MPO peroxidase cycle [31,32]. Most of these studies were based on pre-steady state conditions and/or steady-state conditions with optical spectroscopic monitoring. In most cases, larger than normal plasma concentrations of MPO and/or NO2- were employed. The reason for doing that was to optimize conditions where measurable changes in absorbance at selected wavelengths (430 nm for compound I decay and 455 nm for compound II formation and decay) could be monitored.
Steady-state methods with electrochemical monitoring have advantages over other methods. In such methods a targeted reactant or product could be detected by oxidation or reduction at the surface of a selective electrochemical biosensor. However, lack of biosensors with enough sensitivity, selectivity and short response time limited their employment. Kettle et al. employed a conventional H2O2 electrode for monitoring steady-state loss of H2O2 in MPO-catalyzed reactions with halides, and other one electron reductants [31-35]. Lower sensitivity, long calibration procedure and interferences from produced hypohalous acids limited the ability of collected data from predicting kinetic models. In most cases, initial rates were calculated and employed for either comparison between reductants or as supportive to other methods.
]]> Recently, combination-H2O2 biosensors based on a flexible activated carbon fiber sensing electrode coated with a propriety membrane that enhances H2O2 detection were developed [37,38]. Such electrodes have better sensitivity than conventional electrodes, and a relatively short response time (2s).In this study, we employed a carbon fiber based H2O2-biosensor to study the inhibitory effect of NO2- on catalytic activity of MPO towards oxidation of halides and thiocyanate (Cl-, Br-, I- and SCN-) under respective physiological concentrations. All experiments were based on monitoring the time course decay of the amperometric H2O2 initial signal throughout the MPO-catalyzed reaction. Our results provided comprehensive electrochemical evidence that NO2- inhibits the MPO-catalyzed oxidation of halides and thiocyanate. Observed inhibition was strong for iodide and bromide, and weak for chloride and thiocyanate.
Experimental
Reagents
Chemicals used for preparation of buffer, stock and standard solutions were of analytical grade reagents and purchased from Sigma. Phosphate buffer, 100 mM and pH 7.00, was prepared by mixing appropriate volumes of 0.10 M NaH2PO4 and 0.10 M Na2HPO4 to achieve pH 7.00. A 1.00 mM H2O2 solution was freshly prepared from stock solutions prepared by sequential dilutions from 30 % H2O2 solution. Standard solutions of halides (Cl-, Br-, I-), SCN- and NO2- were prepared by sequential dilutions from their respective sodium salts. All solutions were bubbled with high purity N2 gas before use.
MPO preparation
MPO was purified from detergent extracts from human leukocytes. The purity of isolated MPO was established by demonstrating a Reinheitszahl (RZ) value of > 0.85 (A430/A280) via SDS-PAGE analysis [39-40]. MPO concentration was determined spectrophotometrically utilizing molar extension coefficients of 89.000 and 112.000 M-1 cm-1 per heme (l = 430 nm) [40]. A 30 mM MPO solution was freshly prepared by diluting measured amounts with buffer.
]]> Electrochemical measurements
Amperometric measurements were performed by Apollo 4000 free radical analyzer (WPI, Sarasota, FL, USA). The biosensor was ISO-HPO-100 (also from WPI), that is a 100 μM tip diameter hydrogen peroxide micro sensor. The sensor design was based on a flexible carbon fiber sensing electrode coated with a selective membrane that enhances H2O2 detection. The sensor incorporates combination electrode technology. Applied potential was set at 400 mV. The sensor has sensitivity better than 2 pA/nM, detection limit of 20 nM and a baseline drift of 1 pA/min [38]. Measurements and reactions were monitored in a thermostated measurement chamber (also from WPI) and all experiments were performed at room temperature (25 ºC). The electrode was calibrated daily in presence of 100 µM NO2- and selected physiological concentrations of halides and thiocyanate. Electrode calibrations were based on successive addition of 5 μL from 1.0 mM H2O2 solution to 3.0 mL of 100 mM phosphate buffer solution (pH 7.00) pre-incubated with 100 µM NO2- and a selected physiological concentration of halide or thiocyanate in the chamber. After each addition, the current (nA) was recorded and calibration curves between current (nA) and [H2O2] (μM) were constructed by linear least-squares method. Amperometric monitoring of H2O2 during MPO-catalyzed reaction experiments was performed as follows: for each experiment, 3.00 mL of 100 mM phosphate buffer solution containing 30 μM ethylenediaminetetraacetic acid (EDTA) were placed in the measurement chamber. For blank study measurements, varied concentrations of NO2- (0-100) μM were pre-incubated with buffer. For the effect of nitrite measurements, a selected physiological concentration of halide or thiocyanate (100 mM Cl-, 60 μM Br-, 10 μM I-, 50 μM SCN-) and varied concentrations of NO2- (0-100) μM were pre-incubated with buffer solution in the chamber. For the effect of chloride measurements, 100 μM NO2- and varied concentrations of Cl- (0-100) mM were pre-incubated with buffer solution in the chamber. The electrode was immersed and magnetic stirrer was turned on at fixed moderate speed. Continuous amperometric monitoring started after the addition of 30 μL H2O2 (10 μM). Reactions started after addition of 5.0 μL MPO solution (50 nM) and were allowed to proceed until complete decay of initial current signal. H2O2 concentrations (μM) versus time (s) plots were obtained by setting the initial current signal to 10 μM H2O2.
Results
Validation of measurements
Calibration curves in phosphate buffer solution (blank solution) were linear in the studied range (0.00-10.00 μM H2O2) with a regression equation of Y= (2.21± 0.05) X + (0.11± 0.02) , R2= 0.998, where Y is the current (nA) and X is [H2O2] (μM). Coefficient of variation of slope (CV) is 2.3 % and intercept was equivalent to 0.05±0.01 μM H2O2. Limit of detection (LOD) calculated at S/N= 3, was 0.025 μM. These parameters did not vary significantly (<5 %) upon pre-incubation of blank solution with physiological concentrations of halides, thiocyanate and nitrite. Stability of the electrode was demonstrated by performing calibration measurements daily. Proper use and maintenance of electrode may extend its life up to six months.
Optimization of initial concentrations
The major objective of this work was to conduct a steady-state electrochemical study of the inhibitory effect of nitrite on the catalytic activity of MPO towards oxidation of Cl-, Br-, I- and SCN-. Pre-steady state studies reported that accumulation and stability of compound II by NO2- is the main factor of inhibition of the catalytic reaction [31,32]. It also was documented that H2O2 reacts with compound I to either liberate oxygen gas (catalase) or reduce it to compound II [35,41].Thus, a blank study was conducted to optimize initial MPO and H2O2 concentrations that minimize and estimate the catalase reaction (consumption of H2O2 through oxidation of H2O2 to O2). Additionaly, another study was conducted to select initial halide or SCN- concentration (preferably from physiological range) that when incubated with optimized concentrations of MPO, H2O2 and in absence of nitrite, compound II is not formed during turnover (oxidation of X- to HOX). Results were evaluated by referring to monophasic and biphasic nature of the peroxidation cycle. We assumed that the time course of H2O2 consumption is monophasic when total decay of initial H2O2 signal occurs in < 10 s (equations 1 and 2 in peroxidase cycle). Time course is biphasic when turnover occurs in two sequential and separable steps, a rapid step (first 10 s) followed by a slower step. The first step (fast) is similar to the monophasic step and represents turnover before significant accumulation of compound II. The second step (slow) represents turnover after accumulation of compound II (partial conversion of MPO to compound II, equations 1-3 in peroxidase cycle).
Blank decay plots were biphasic. Rates of consumption of H2O2 from both steps increase by increasing MPO and H2O2 concentrations due to dominance of catalase reaction [35]. A typical decay plot at selected H2O2 (10 μM ) and MPO (50 nM) is presented in Fig. 1 (a). Blank decay plots did not change upon incubation of blank samples with NO2- (up to 100 μM) and with 10 μM of hypohalous acid. Calculated rates from blank studies were 0.10 μM/s for first step and 0.03 μM/s for second step. Pre-incubation of blank samples with increasing concentrations of halides or SCN- (absence of NO2-) has increased both rates and decay plots were monophasic at Cl-> 15 mM, Br-> 50 μM, I-> 5.0 μM and SCN- > 40 μM (data not shown). Selected initial concentrations, 100 mM Cl-, 60 μM Br-, 10 μM I- and 50 μM SCN- were from physiological range except for I- (plasma range ~ 1.0 μM) due to stoichiometric reasons (same concentration of H2O2).
]]>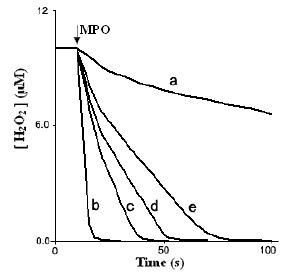
Figure 1. Effect of NO2 - on MPO-catalytic activity towards oxidation of Cl-. H2O2 consumption plots as a function of NO2 -. Reactions were started by the addition of 50 nM MPO to 10 µM H2O2 in 100 mM phosphate buffer, pH 7.0, containing 30 µM EDTA (a) pre-incubated with 100 mM Cl- (b) and 25 (c), 50 (d), 100 µM NO2 - (e). Reactions were arried at 25 oC. Plots are average of four replicates.
Inhibitory effect of NO2- on MPO-catalytic activity towards oxidation of Cl-
Chloride is assumed to be the physiological substrate for MPO due to its high concentration (100-140 mM), in contrast to 20-100 μM bromide, 0.1-0.6 μM iodide and 20-120 μM thiocyanate. Time course H2O2-decay plots for MPO-catalyzed oxidation of Cl-, at a selected normal plasma level (100 mM), in presence of increasing NO2-concentrations were examined by continuous amperometric monitoring of H2O2 consumption (Fig. 1 (b-e)). Monophasic plots prevail for NO2- concentrations 0-20 μM, accompanied with a drop of rate of consumption of H2O2 from 1.5 to 0.75 μM/s. As NO2- concentrations exceed 25 μM/s, plots became biphasic and accompanied with further decrease of consumption rates from both steps. Rate results and reaction times are summarized in Table 1.
Table 1. Relative rates and reaction times for MPO-catalyzed reaction of hydrogen peroxide with selected physiological concentrations of halides and thiocyanate, at varied concentrations of nitrite*.
Results in Table 1 show that NO2- inhibited both rapid and slow steps. Rates of first step were reduced from 1.5 (absence of nitrite) to 0.30 μM/s (100 μM NO2-), while rates of second step were reduced from 0.30 (25 μM NO2-) to 0.13 to μM/s (100 μM NO2-). Total H2O2-consumption time increased from 6 to 60 s. Since lower rate values are larger than blank values (0.1 and 0.03 μM/s), nitrite is considered a weak inhibitor to MPO catalytic activity towards oxidation of chloride.
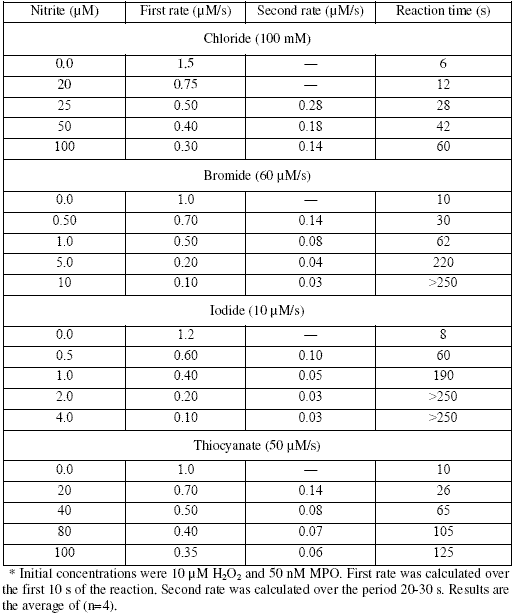
To study the effect of Cl- concentration on NO2- inhibition of the reaction we pre-incubated the reaction with 100 μM NO2- and varied concentrations of Cl-. Time course plots of consumption of H2O2 were biphasic and similar to those in Fig. 1 with a decrease in both rates by decreasing Cl- concentration; rate results and reaction times are summarized in Table 2. Total H2O2-consumption time increased from 60 s to 240 s. At 5 mM Cl- (20 times lower than normal plasma concentration) and 100 μM NO2- (25 times lower than normal plasma concentration) calculated rates were 0.12 and 0.04 μM/s from first and second steps, respectively. Since, these values are similar to blank values we assume that complete inhibition could occur under conditions far from physiological concentrations of Cl- and NO2-.
Table 2. Relative rates and reaction times for MPO-catalyzed reaction of hydrogen peroxide with varied concentrations of chloride, at 100 μM nitrite*.
Effect of NO2- on MPO-catalytic activity towards oxidation of Br-, I-and SCN-
Even bromide and iodide are not preferred substrates for MPO, their MPO catalyzed oxidation was investigated [42]. Also, it was reported that SCN- is a preferred substrate for MPO and its MPO catalyzed reaction was extensively studied [34,42]. Thus, we extended our study to include for the first time bromide, iodide and thiocyanate.
Initial decay plots (absence of nitrite) were monophasic. As NO2- concentration increased reactions became biphasic. Calculated rates and total H2O2-consumption times are summarized in Table 1. For bromide, the second step was completely inhibited at 5.0 μm NO2- and both steps were completely inhibited at 10 μm NO2-. For iodide, the second step was completely inhibited at 2.0 μm NO2- and both steps were completely inhibited at 4.0 μm NO2-. These results imply that nitrite is a strong inhibitor to their MPO catalyzed oxidation reactions. In contrast to bromide and iodide, nitrite is a weak inhibitor to MPO catalyzed oxidation of thiocyanate. At 100 μm NO2- (double of initial concentration of SCN-) rates of consumption of H2O2 were 0.35 and 0.06 μm/s from both steps, respectively.
]]>Discussion
Our major objective in this study was to explore the importance of electrochemical measurements in enzyme kinetics. We acknowledge that other non-electrochemical measurements are still required to reach a complete kinetic model.
Assessment of MPO-catalytic activity towards oxidation of halides and pseudo halides is a complex and multifunctional process [24,37]. MPO catalytic activity is dependent on initial concentrations of MPO, H2O2 and halide or pseudo halide, H2O2 to MPO concentration ratio and order of addition.
Because MPO compound I formation rate is slower than the 2e oxidation of halides or thiocyanate, compound I cannot be detected during steady state catalysis [20,44,45]. Thus, the rate of formation and accumulation of compound II will be a measure of decrease in MPO catalytic activity.
In this study, we presented a comprehensive electrochemical evidence that nitrite inhibits the MPO catalytic activity towards oxidation of halides and thiocyanate. Our results show for the first time that nitrite is a strong inhibitor for oxidation of iodide and bromide and a weak inhibitor for oxidation of thiocyanate and chloride.
Burner et al. studied the mechanism of reaction of MPO with nitrite [32]. They investigated the reaction of compound I and compound II with nitrite under pre-steady state conditions by using sequential mixing stopped-flow measurements. They concluded that nitrite is oxidized by two one-electron steps in the MPO peroxidase cycle.
The second-order rate constant of reduction of compound I to compound II (Eq. 5) by nitrite is 2.0 x 106 M-1s-1 and reduction of compound II to native MPO (Eq. 6) is 5.5 x 102 M-1s-1. These results indicate that nitrite inhibits the reaction by formation and accumulation of compound II.
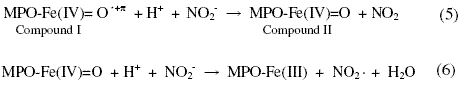
Other pre-steady state studies with stopped-flow monitoring were conducted to study MPO catalytic activity in presence of chloride, bromide, iodide and thiocyanate [23, 25, 35, 42-45]. Reported second-order rate constants varied from 1 x 107 to 4.3 x 107 M-1s-1 for formation of compound I. Second-order rate constants for two electron oxidation of halides and thiocyanate by compound I were from 2.5 x 104 (physiological concentration) to 4.7 x 106 (lower concentration) for chloride, 1.1 x 106 M-1s-1 for bromide, 7.2 x 106 M-1s-1 for iodide and 9.6 x 106 M-1s-1 for thiocyanate. Second-order rate constant for formation of compound II from reaction of compound I and hydrogen peroxide was from 3.5 x 104 to 8.2 x 104 M-1s-1. Variations between reported results are expected and are mainly attributed to initial conditions and advance of employed instrumentation. Thus, for steady-state studies of inhibition effect of nitrite on MPO catalytic activity towards oxidation of halides and thiocyanate, initial concentrations should be optimized that, in absence of nitrite, complete decay of initial H2O2 signal should occur without any accumulation of compound II.
Increasing concentrations of nitrite inhibits the MPO catalyzed oxidation of halides and thiocyanatate by initially decreasing the rate of initial rapid step followed by altering the reaction to biphasic and in-sequence decreasing rates of both rapid and slow steps.
Inhibition effect of NO2- on MPO catalytic activity was observed in all studied halides and thiocyanate. Inhibition increases upon increasing nitrite concentration and was largest for iodide and bromide. Complete inhibition occurred at nitrite concentrations of 4.0 μM for iodide, 10 μM for bromide and partial inhibition was observed for chloride and thiocyanate even at 100 μM nitrite.
Very high physiological concentration of chloride relative to other halides and thiocyanate has contributed to this effect. Complete inhibition occurred approximately at 5.0 mM Cl- (twenty times lower than physiological level) and 100 μM NO2- (twenty five times larger than physiological concentration) (Table 2). Thiocyanate and bromide have approximately the same physiological levels and rates of 2e oxidation by compound I. Thus, thiocyanate is expected to follow bromide of being strongly inhibited by nitrite. It was documented that thiocyanate modulates catalytic activity of MPO by being oxidized via 2e and two 1e mechanisms. Thiocyanate reduces compound II to ground state MPO with a second-order rate constant of 1 x 104 M-1 s-1 (eq. 7) [24].
![]()
A general kinetic scheme describing how NO2- inhibits the MPO-catalyzed oxidation of halides and thiocyanate is illustrated in Fig. 2. In absence of nitrite, initial conditions were optimized that complete decay of H2O2 occurs through 2e oxidation without formation of compound II (enzyme is swinging between compound I and native MPO and working in maximum activity) which was represented by the initial rapid and monophasic step (< 10 s). In presence of increasing concentrations of nitrite, part of compound I is reduced to compound II (Eq. 5) by causing the enzyme to work at fraction of maximum activity, which was represented by the biphasic character of the amperometrically monitored H2O2-decay signal.
]]>
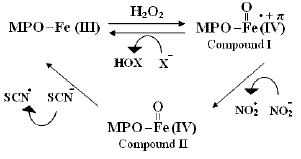
Figure 2. Simple kinetic model representing the inhibitory effect of NO2-.
Conclusions
A reliable electrochemical method has been employed for the study of inhibitory effect of nitrite on MPO-catalytic activity towards oxidation of halides and thiocyanate. Sensitivity of the method was demonstrated by presenting the monophasic and biphasic character of the amperometrically monitored H2O2-decay plots and the ability to distinguish between two different rates in the first 30 s of the reaction. Selectivity of the method was verified by monitoring H2O2-decay plots with reaction times between 6 s and > 250 s without significant interferences from pre-incubated reactants and produced hypohalous acids.
Our results demonstrated that under physiological concentrations of halides and thiocyanate, nitrite is a strong inhibitor to MPO-catalyzed oxidation of iodide and bromide. Nitrite is a weak inhibitor of MPO-catalyzed oxidation of chloride and thiocyanate, which is largely due to high physiological concentration of chloride and reduction of compound II to native MPO by thiocyanate.
Limitations of electrochemical measurements in enzyme kinetics are mainly due to relatively long response time of available electrodes (seconds). For example, in our study the rapid step (first 10 s of the reaction) needs to be resolved and this could be done by modifying response time of H2O2-biosensor to milliseconds rather than seconds. Additionally, the development of MPO-biosensor will solve the puzzle of MPO partitioning between native enzyme and intermediates during steady state catalysis. The ultimate goal of success of electrochemical measurements in enzyme kinetics is the ability to perform pre-steady state catalysis with electrochemical detection.
Acknowledgments
This work was done at Department of Obstetrics and Gynecology, School of Medicine, Wayne State University, Detroit, MI, USA. The principal author would like to thank Jordan University of Science and Technology for financing research sabbatical to conduct this study.
]]>References
1. S. Galijasevic, G. Saed, M. Diamond, H. Abu-Soud, PNAS 100 (2003) 14766. 10.1073/pnas.2435008100
2. L. Ignarro, Ann. Rev. Pharmacol. Toxicol. 30 (1990) 535. 10.1146/annurev.pa.30.040190.002535
3. S. Moncada, R. Palmer, E. Higgs, Pharmacol. Rev. 43 (1991) 109. [ Links ]
4. S. Vincent, Prog. Neurobiol. 42 (1994) 129. 10.1016/0301-0082(94)90023-X
5. E. Anggard, Lancet 343 (1994) 1199. 10.1016/S0140-6736(94)92405-8![]()
6. J. Beckman, W. Keppenol, Am. J. Physiol. 271 (1996) C1424.
7. L. Ignarro, J. Fukuto, J. Griscavage, N. Rogers, R. Byrns, Proc. Natl. Acad. Sci. USA 90 (1993) 8103.
8. D. Torre, G. Ferrario, F. Sperenza, A. Orani, G. Fiori, C. Zeroli, J. Clin. Path. 49 (1996) 574. 10.1136/jcp.49.7.574
]]> 9. A. Farrell, D. Blake, R. Palmer, S. Moncada, Am. Rheum. Dis. 51 (1992) 1219. 10.1136/ard.51.11.1219.10. A. Leone, P. Francis, R. Rhodes, S. Monkada, Biochem. Biophys. Res. 200 (1994) 951. 10.1006/bbrc.1994.1542
11. T. Ueda, T. Maekawa, D. Sadamitsu, S. Oshita, K.Ogino, K. Nakamura, Electrophoresis 16 (1995) 1002. 10.1002/elps.11501601167
12. B. Gaston, J. Reilly, J. Drazen, J. Fackter, P. Ramdey, D. Arnelle, M. Mollins, D. Sugrabaker, C. Chee, D. Singel, J. Loscalazo, J. Stamler, Proc. Natl. Acad. Sci. USA 90 (1993) 10957.
13. L. Green, D. Wagner, J. Glokowiski, P. Skipper, J. Wishnok, S. Tannenbaum, Anal. Biochem. 126 (1982) 131. 10.1016/0003-2697(82)90118-X
14. S. Klebanoff, J. Leukocyte Biol. 77 (2005) 598. 10.1189/jlb.1204697
15. M. Belding, S. Klebanoff, G. Ray, Science 167 (1970) 195. 10.1126/science.167.3915.195
16. S. Kimura, M. Ikeda-Saito, Proteins 3 (1988) 113.
17. E. Jong, W. Henderson, S. Klpanoff, J. Immunol. 124 (1980) 1378.
18. S. Weiss, S. Test, C. Eckman, D. Roos, S. Regiani, Science 234 (1986) 200. 10.1126/science.3018933
]]> 19. S. J. Klebanoff, A. M. Waltersdroph, H. Rosen, Methods Enzymol. 105 (1984) 399. 10.1016/S0076-6879(84)05055-220. A. J. Kettle, C. C. Winterbourn, Redox. Rep. 3 (1997) 89-107.
21. J. Hurst, in Peroxidases in Chemistry and Biology, J. Everse, K. Everse, M. Grisham, Eds., Vol. 1, CRC. Press, Boca Raton-Fl, 1991. p. 37.
22. L. Marquez, J. Huang, H. Hunford, Biochemistry 33 (1994) 1447. 10.1021/bi00172a022
23. L. Marquez, H. Dunford, H. van Wart, J. Biol. Chem. 265 (1990) 5666.
24. Y. Tahboub, S. Galijasevic, M. Diamond, H. Abu-Soud, J. Biol. Chem. 280 (2005) 26129. 10.1074/jbc.M503027200
25. S. Galijasevic, I. Abdulhamid, H. Abu-Soud, Free Radic. Biol. Med. 44 (2008) 1570. 10.1016/j.freeradbiomed.2008.01.003
26. H. Shibata, Y. Kono, S. Yamashita, Y. Sawa, H. Ochiai, K. Tanaka, Biochim. Biophys. Acta 1230 (1995) 45. 10.1016/0005-2728(95)00031-D
27. A. vander Vliet, J. Eiserich, C. O'Neill, B. Halliwell, C. Cross, Arch. Biochem. Biophys. 319 (1995) 341. 10.1006/abbi.1995.1303
28. E. Monzani, R. Roncone, M. Galiano, W. Koppenol, L. Casella, Eur. J. Biochem. 271 (2004) 895. 10.1111/j.1432-1033.2004.03992.x
]]> 29. A. vander Vliet, J. Eiserich, B. Halliweel, C. Cross, J. Biol. Chem. 272 (1997) 7617. 10.1074/jbc.272.12.761730. J. Eiserich, C. Cross, A. Jones, A. vander Vliet, J. Biol. Chem. 271 (1996) 19199. 10.1074/jbc.271.32.19199
31. C. van Dalen, C. Winterbourn, R Santhilmohan, A. Kettle, J. Biol. Chem. 275 (2000) 11638. 10.1074/jbc.275.16.11638
32. U. Burner, P. Furtmuller, A. Kettle, W. Koppenal, C. Obinger. J. Biol. Chem. 275 (2000) 20597. 10.1074/jbc.M000181200
33. A. Kettle, C. Winterbourn, Methods Enzymol. 233 (1994) 502. 10.1016/S0076-6879(94)33056-5
34. C. van Dalen, M. Whiterhouse, C. Winterbourne, A. Kettle, Biochem. J. 327 (1997) 487.
35. A. Kettle, C. Winterbourne, Biochemistry 40 (2001) 10204. 10.1021/bi010940b
36. J. Wang, Electroanalysis 17 (2005) 7. 10.1002/elan.200403113
38. R. Rakita, R. Michel, H. Rosen, Biochemistry 29 (1990) 1075. 10.1021/bi00456a033
]]> 39. R. Wever, H. Plat, M. Hamers, FEBS Lett. 123 (1981) 327.40. K. Agner, Acta. Chem. Scand. 17 (1963) S332.
41. H. Hoogland, H. Dekker, C. van Riel, A.van Kuilenburg, A. Muijsers, R. Wever, Biochimica et Biophysica Acta 955 (1988) 337. 10.1016/0167-4838(88)90213-0
42. P. Furtmuller, C. Obinger, Y. Hsuanyu, H. Dunford, Eur J. Biochem. 267 (2000) 5858. 10.1046/j.1432-1327.2000.01491.x
43. P. Furtmuller, U. Burner, C. Obinger, Biochemistry 37(1998) 17923. 10.1021/bi9818772
44. H. Dunford, Y. Hsuanyu, Biochem. Cell Biol. 77 (1999) 449. 10.1139/bcb-77-5-449
45. J. Arnold, P. Furtmuller, C. Obinger, Redox Rep. 8 (2003) 179. 10.1179/135100003225002664
Received 8 June 2009; accepted 15 December 2009
]]> * Corresponding author: tahboub@just.edu.jo ]]>