Neurofibromatose tipo 1: relato de um caso clínico
Neurofibromatosis type 1: a case report
Ana Catarina Marques*, Fátima Dinis**
*Interna de 3.o ano de formação específica em Medicina Geral e Familiar
Unidade de Saúde Familiar Fafe Sentinela - ACES do Alto Ave
**Interna de 2.o ano de formação específica em Medicina Geral e Familiar
Unidade de Saúde Familiar de Ronfe - ACES do Alto Ave
Endereço para correspondência | Dirección para correspondencia | Correspondence
]]> RESUMO
Introdução: A neurofibromatose tipo 1 (NF1) é uma doença neurocutânea de hereditariedade autossómica dominante, ocorrendo mutações de novo em cerca de metade dos casos. Sendo o diagnóstico firmado na presença de pelo menos dois critérios de diagnóstico, na maioria clínicos, o médico de família deve conhecer as manifestações desta doença.
Caso: AIGD, sexo feminino, 29 anos, com história de numerosas lesões cutâneas desde criança, de agravamento progressivo e dificuldades de aprendizagem. Antecedentes maternos de lesões cutâneas semelhantes. Pertence a uma família alargada, de classe média-baixa, com critérios de risco familiar. Em 2008 foi referenciada a consulta de Cirurgia Geral pelo seu médico de família por «múltiplos nevos e verrugas». Após a exérese de várias destas lesões, foi feito o diagnóstico anatomopatológico, em 2009, de neurofibromas. Foi enviada para consulta de Neurologia, na qual se confirmou apresentar critérios clínicos para diagnóstico de NF1 (neurofibromas, efélides axilares, ≥ seis manchas cutâneas de cor café com leite). Nessa consulta havia registo de um filho de 3 anos com manchas cutâneas de cor café com leite. A criança foi convocada para observação por Dermatologia apenas em 2012, após observação da utente por esta especialidade. Nesta consulta, verificou-se que o filho apresentava também critérios para diagnóstico de NF1.
Comentário: A NF1, pela sua frequência (1/2500-3000) e gravidade das complicações associadas, constitui uma doença cujo diagnóstico precoce é importante. Como é evidente neste caso, nem sempre os médicos estão atentos às suas manifestações clínicas, perdendo-se a oportunidade de aconselhamento genético e/ou de minimizar as complicações desta doença.
Palavras-Chave: Neurofibromatoses; Neurofibromatose 1; Neurofibromas.
ABSTRACT
Introduction: Neurofibromatosis type 1 (NF1) is a common neurocutaneous condition with an autosomal dominant pattern of inheritance. A new mutation occurs in about half of the cases. The diagnosis is based on clinical assessment so family physicians should be aware of the manifestations of the disease.
Case presentation: A 29 year-old female patient presented with a history of numerous skin lesions since childhood. The lesions were getting progressively worse. The patient also had learning disabilities. Her mother had similar skin lesions. She was referred to a General Surgery clinic in 2008 because of multiple moles and warts. After excision of skin lesions, a pathological diagnosis, of neurofibromas was made in 2009. She was referred to a Neurology clinic and the clinical diagnosis of NF1 was confirmed by the presence of neurofibromas, freckles, and more than 6 café au lait spots on the skin. It was also noted that her three year-old son had one café au lait patch on his skin. The child was referred to a Dermatology clinic in 2012. At this time the child fulfilled the clinical criteria for the diagnosis of NF1.
Commentary: Because of the prevalence (1/2500-3000) and severity of complications of NF1, the early detection of the disease is important. As is evident in this case, clinicians are not always aware of its manifestations, missing opportunities for genetic counselling and preventing complications of the disease.
]]> Key-Words: Neurofibromatoses; Neurofibromatosis 1; Neurofibroma.
Introdução
Aneurofibromatose tipo 1 (NF1) é uma doença neurocutânea, cuja incidência estimada é de 1 em cada 2500-3000 indivíduos.1-3 Os primeiros relatos desta doença datam do ano 1000 aC.1 No entanto, só em 1881 von Recklinghausen descreveu e apelidou de «neurofibromas» os tumores benignos que cresciam da bainha dos nervos periféricos; a NF1 é, ainda hoje, alternativamente designada por «Doença de von Recklinghausen».1 Em 1990 foi identificado o gene no qual ocorre a mutação responsável pela doença – o gene NF1 do cromossoma 17q11.2. Na presença de mutação deixa de haver síntese de neurofibromina, um supressor tumoral com expressão ubiquitária no sistema nervoso, condicionando um risco significativamente aumentado de desenvolvimento de tumores benignos e malignos por todo o sistema nervoso.1
A propósito de um caso clínico
Identificação
A.I.G.D., sexo feminino, 29 anos, portuguesa*. Completou o 6.o ano de escolaridade. Atualmente desempregada. Vive em união de facto. Reside, desde 2007, com o filho de 5 anos de idade e com o companheiro em casa dos pais deste, pertencendo assim a uma família alargada. A família enquadra-se na classe IV da classificação de Graffar (classe média-baixa) e apresenta critérios de risco familiar médio, segundo as escalas de Segovia-Dreyer e de Garcia Gonzalez.
*Foram omitidos alguns dados para melhor proteção da identidade da utente.
Antecedentes pessoais e familiares
Trata-se de uma utente sem médico de família atribuído, que mantém contacto esporádico na Unidade de Saúde Familiar (USF)2 onde o filho se encontra inscrito.
]]> Tem antecedentes pessoais de dificuldade de aprendizagem e numerosas lesões cutâneas (pápulas e nódulos de pequenas dimensões) desde criança, que foram aumentando em número e tamanho com o decorrer da idade.Como antecedentes familiares de relevo tem mãe com lesões cutâneas semelhantes.
História da doença atual
Em janeiro de 2006, com 23 anos de idade, foi referenciada à consulta de Obstetrícia do hospital de referência pelo Centro de Apoio a Toxicodependentes (CAT), ao qual recorreu pela primeira vez por teste de gravidez positivo após relação sexual desprotegida com namorado, seropositivo para o Vírus da Imunodeficiência Humana (VIH).
Foi seguida em consulta hospitalar de gravidez de risco, tendo-se verificado sempre seronegatividade da utente para o VIH 1 e 2. Não foram detetados outros fatores de risco. O parto decorreu em setembro de 2006, sem intercorrências.
Por agravamento das lesões cutâneas após a gravidez, em número e tamanho, a utente realizou um contacto esporádico na USF em novembro de 2007. Nesta consulta, foi constatada a presença de várias lesões cutâneas, descritas como «múltiplas verrugas na pele de distribuição generalizada», pelo que foi feita a referenciação para consulta de Cirurgia Geral.
Na primeira consulta de Cirurgia Geral, em fevereiro de 2008, observaram-se ao exame objetivo «três pápulas no antebraço direito, antebraço esquerdo e coxa esquerda». Os antecedentes pessoais e familiares foram considerados irrelevantes. Em agosto de 2008 foi efetuada exérese das lesões descritas, que histologicamente correspondiam a nevos melanocíticos intradérmicos.
Na consulta de reavaliação, em fevereiro de 2009, a utente apresentava novas lesões, descritas como «um quisto sebáceo, uma pápula fibrosa e seis pápulas nos membros». Em maio de 2009 foi realizada a exérese destas lesões, cujo exame anatomopatológico revelou «neurofibromas». Foi referenciada em junho de 2009 para consulta de Neurologia, por «clínica compatível com neurofibromatose».
Na primeira consulta de Neurologia, que se realizou em outubro de 2009, aos 26 anos de idade, foi confirmada a presença de «múltiplos quistos sebáceos, neurofibromas de vários tamanhos, manchas café com leite e efélides». Não existia história de epilepsia, cefaleias ou alterações ao exame neurológico. Apresentava avaliação tensional normal. Foram registados nesta consulta, como antecedentes familiares de relevo, «mãe com o mesmo problema de pele» e «filho de 3 anos com uma mancha café com leite». Foi pedida Ressonância Magnética Nuclear (RMN) cerebral e marcada consulta de seguimento.
Em novembro de 2010, a doente mantinha-se sem queixas e a RMN cerebral, realizada em março de 2010, revelou «suspeita de hamartoma a nível do vérmis, hipersinal talâmico punctiforme direito no mesmo contexto e discreta assimetria da hipófise com normal impregnação».
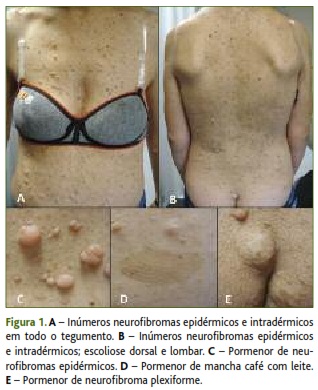
]]> Foi referenciada a Dermatologia, para avaliação da necessidade de exérese de algumas lesões/quistos, tendo a primeira consulta sido realizada em novembro de 2011. Apresentava então «incontáveis neurofibromas epidérmicos e intradérmicos em todo o tegumento, efélides de localização axilar e inguinal, mais de seis manchas café com leite» e pectus excavatum (Figura 1). Por reunir critérios clínicos para diagnóstico de NF1, foi pedida avaliação por Oftalmologia, radiografia da coluna e ossos longos, ecografia abdominal e renovesical, e recomendada vigilância periódica da tensão arterial.
Na avaliação oftalmológica foi detetado erro de refração, corrigido com óculos. Não foram detetadas outras alterações, nomeadamente a presença de hamartoma da íris (nódulos de Lisch), e foi recomendada vigilância periódica.
A radiografia da coluna vertebral evidenciava «segmento dorsal com atitude escoliótica dorsal de concavidade esquerda e segmento lombar com escoliose lombar de concavidade direita».
Nesta consulta de Dermatologia foi convocado o filho da utente.
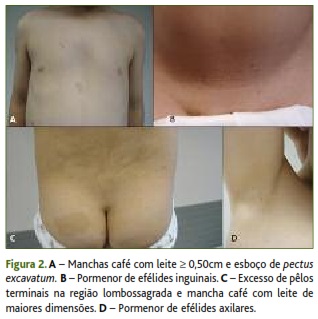
Na primeira observação da criança, em junho de 2012, eram evidentes seis manchas café com leite ≥ 0,50 cm, efélides axilares e inguinais, esboço de pectus excavatum e excesso de pêlos terminais na região lombossagrada (Figura 2). Confirmou-se assim a presença de critérios clínicos suficientes para o diagnóstico de NF1 e foi feita referenciação para avaliação multidisciplinar.
]]>
Acerca da NF1
As complicações da NF1 são diversas e a expressão da doença varia, mesmo dentro da mesma família.1,2,4 É transmitida por hereditariedade autossómica dominante, apesar de em aproximadamente metade dos doentes não existir história familiar (mutações de novo).1
A NF é classificada como um síndrome de predisposição tumoral, pela incidência aumentada de tumores benignos e malignos.5 Os doentes com NF têm uma diminuição da esperança média de vida de cerca de 15 anos. A principal causa de morte em adultos jovens são os tumores malignos das bainhas nervosas periféricas, que são muito agressivos e com elevada capacidade de metastização, podendo ocorrer em vários locais do sistema nervoso periférico. Os neurofibromas podem sofrer transformação maligna numa proporção três a cinco vezes superior à da população não afetada pela NF.1,5
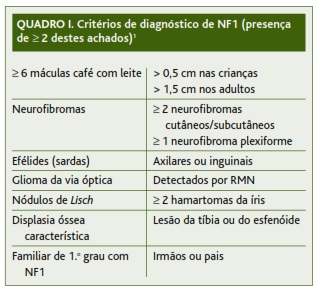
O diagnóstico da doença é frequentemente firmado em características clínicas, sendo estabelecido na presença de pelo menos dois dos critérios de diagnóstico descritos no Quadro I.1-3 Como tal, para que este diagnóstico possa ser precoce, é necessário que o clínico esteja alerta para as primeiras manifestações da doença.
As primeiras manifestações da doença são cutâneas, usualmente sob a forma de manchas café com leite. Uma a duas manchas café com leite ocorrem em até 10% da população geral, sem que isso tenha significado patológico.1 No entanto, a presença de três ou mais manchas, principalmente se presentes desde a infância e mesmo sem que existam antecedentes familiares, deve deixar o clínico alerta para a possível existência de NF1.1 As efélides axilares e inguinais são igualmente frequentes e caraterísticas desta patologia e, como tal, devem ser ativamente pesquisadas. Os neurofibromas surgem habitualmente mais tarde, no entanto, por serem tão caraterísticos da NF1, constituem um auxiliar precioso para o diagnóstico.1-4 A estimulação hormonal durante a puberdade e gravidez conduz ao agravamento das lesões pré-existentes e ao desenvolvimento de novas lesões.5
Das manifestações musculoesqueléticas, a escoliose é a mais frequente, podendo afetar 20% dos doentes e associar-se a neurofibromas paravertebrais. A baixa estatura e incidência de fraturas são superiores nos doentes com NF1. Estas manifestações parecem dever-se à diminuição da concentração sérica de 25-hidroxi-vitamina D, caraterística destes doentes.6,7
]]> Perante achados clínicos sugestivos de NF1, é essencial recolher toda a informação possível acerca das lesões existentes (idade de aparecimento e eventual modificação com a idade, entre outros), averiguar antecedentes familiares (questionando a presença de lesões caraterísticas nos familiares de 1.o grau) e proceder a um exame físico completo e orientado para a pesquisa de outras manifestações da NF1.1-4A requisição de teste genético para pesquisa da mutação caraterística ou a biopsia dos neurofibromas para confirmação anatomopatológica não são habitualmente necessárias. Se os dados clínicos existentes forem sugestivos, mas não suficientes para o diagnóstico de NF1, recomenda-se a vigilância, de periodicidade anual, por parte do médico assistente.1-4
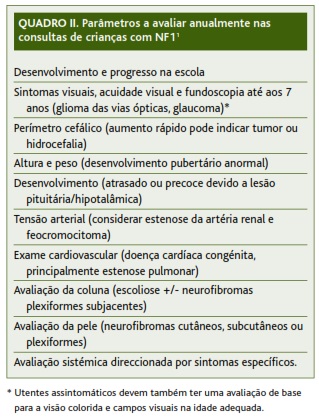
Se as manifestações clínicas forem suficientes para se estabelecer o diagnóstico de NF1, é recomendada a referenciação do utente às especialidades de Dermatologia, Neurologia, Pediatria, Oftalmologia, Genética Médica ou outras, conforme o exame clínico ou estudo complementar assim o indique. O seguimento por parte do médico de família deve ser mantido, com periodicidade mínima anual. Nestas consultas deve ser efetuada uma avaliação cuidadosa, procurando lesões de novo ou evidência de atingimento de outros órgãos (ver Quadro II). O utente com NF1 deve ser esclarecido acerca do diagnóstico, etiologia, complicações e prognóstico da sua doença, e deve ser instruído a recorrer ao médico sempre que surja um sintoma de novo.1-3
O aconselhamento genético deveria ser proposto a todos os portadores de neurofibromatose pois, ao tratar-se de uma doença hereditária autossómica dominante, existe 50% de risco de transmissão da doença. É possível evitar a transmissão da doença através da fertilização in vitro, com pesquisa e seleção dos embriões sem mutação.1
O atingimento da NF1 é multiorgânico, podendo ocorrer nos mais diversos sistemas. No entanto, não está recomendada a requisição de exames auxiliares de diagnóstico para detetar complicações assintomáticas.
Atuação do MF na NF1
A NF1, pela sua frequência e gravidade das complicações a ela associadas, constitui uma doença cujo diagnóstico precoce é de primordial importância.
]]> O médico de família, ao constituir o primeiro contacto do utente com os cuidados de saúde, está numa posição privilegiada para suspeição clínica, diagnóstico e referenciação atempada para os cuidados de saúde hospitalares.Com o diagnóstico tardio, perde-se a oportunidade de deteção, seguimento e controlo eficaz das complicações. Neste caso clínico, a utente apresenta escoliose, que não teve aconselhamento direcionado. Além disso, o diagnóstico tardio não permitiu evitar a transmissão da doença aos seus descendentes.
A convocatória do filho da utente à consulta foi também tardia. Neste caso, decorreram três anos desde a informação de que a utente tinha um filho que apresentava uma mancha café com leite até à sua observação. Este facto condicionou o acesso da criança ao rastreio das complicações mais precoces (avaliação oftalmológica e auditiva, óssea e do desenvolvimento psicomotor) assim como aos cuidados antecipatórios essenciais (proteção solar, programas de educação pré-escolar e terapia da fala, natação e outros exercícios de correção postural, ).
O médico de família tem a oportunidade de, pelo carácter holístico da sua abordagem e pela acessibilidade às suas consultas, realizar um acompanhamento longitudinal destes utentes e respetivas famílias, complementando a consulta hospitalar. Seria de esperar que o filho da utente, nas consultas na USF, possuindo lesões características em número suficiente para se estabelecer o diagnóstico de NF1, tivesse sido diagnosticado e orientado mais precocemente.
O facto de a utente não ter médico de família atribuído pode ter contribuído para o atraso no diagnóstico e orientação. Tal facto vem alertar para a importância de se valorizarem os contactos esporádicos nos Cuidados de Saúde Primários para a realização de uma avaliação global e cuidada, mantendo um olhar atento sobre os achados clínicos encontrados, pesquisando antecedentes familiares e/ou pessoais e não adiando as referenciações pertinentes.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Ferner R, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007 Feb; 44 (2): 81-8. [ Links ]
2. Neurofibromatosis. NIH Consens Statement Online 1987 Jul 13-15; 6 (12): 1-19. [ Links ]
3. Radtke H, Sebold CD, Allison C, Haidle JL, Schneider G. Neurofibromatosis type 1 in genetic counseling practice: recommendations of the National Society of Genetic Counselors. J Genet Couns 2007 Aug; 16 (4): 387-407. [ Links ]
4. Fauci AS, Braunwald E, Casper DL, Hauser SL, Longo DL, Jameson JL, Loscalzo J. Harrison´s Principles of Internal Medicine. 17th ed. New York: McGraw-Hill Professional; 2008. [ Links ]
5. Soares S, Oliveira G, Teixeira Gomes J, Bom Sucesso M, Teixeira Gomes R. Neurofibromatose tipo 1, uma evolução maligna em idade pediátrica. Acta Med Port 2010 Mai-Jun; 23 (5): 515-20.
6. Souza JF, Toledo LL, Ferreira MC, Rodrigues LO, Rezende NA. Neurofibromatose tipo 1: mais comum e grave do que se imagina. Rev Assoc Med Bras 2009 jul-ago; 55 (4): 394-9. [ Links ]
7. Pasmant E, Vidaud M, Vidaud D, Wolkenstein P. Neurofibromatosis type 1: from genotype to phenotype. J Med Genet 2012 Aug; 49 (8): 483-9. [ Links ]
]]>Endereço para correspondência | Dirección para correspondencia | Correspondence
Fátima Alexandra Ferreira Dinis
Rua Antero Henriques da Silva, n.o 389, R/c dto. 4810-026 Costa, Guimarães
Agradecimentos
À Dra. Olga Pereira, orientadora do estágio hospitalar de Dermatologia, pelo apoio prestado na elaboração do presente artigo.
Conflito de interesses
As autoras declaram não ter conflito de interesses.
]]>Recebido em 25/10/2012
Aceite para publicação em 29/09/2013
Artigo escrito ao abrigo do novo acordo ortográfico.
]]>