










Serviços Personalizados
Journal

Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares em
SciELO
Similares em
SciELO Compartilhar

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
versão impressa ISSN 0872-0169
Port J Nephrol Hypert vol.26 no.4 Lisboa out. 2012
Hereditary hypophosphataemic rickets: experience from a paediatric nephrology unit
Joana S. Caetano, Carolina Cordinhã, Clara Gomes, António J. Correia
Hospital Pediátrico Carmona da Mota, Coimbra, Portugal.
ABSTRACT
Introduction. Rickets is a paediatric disease which should be suspected in children presenting with failure to thrive, motor developmental delay and orthopaedic abnormalities. Although rare, hereditary hypophosphataemic rickets is the most common form of heritable rickets.
Patients and Methods. Retrospective observational study of all children with hypophosphataemic rickets observed at a paediatric nephrology unit of a tertiary paediatric hospital from 1982 to 2012, identified from the units database. Data collected included demographics, risk factors, pre-existing medical conditions, clinical, radiographic and laboratory findings, treatment and morbidity.
Results. Eleven children with hypophosphataemic rickets were studied, with a median age at admission of 4.25 years (0.66-10.92). Family history of rickets or orthopaedic abnormalities was found in five children.
The first clinical manifestations were delayed/abnormal gait (7/11) and short stature (4/11). Skeletal deformities were present in all children: genus valgum or varum (11/11), thickening of the wrists (7/11), rachitic rosary (4/11), frontal skull bossing (2/11), Harrisons groove (1/11). Dental abscess was reported in one child and joint pain in six. Laboratory findings included increased alkaline phosphatase (11/11), low serum phosphorus (11/11), normal serum calcium (10/11) and parathyroid hormone values (6/11) and low renal phosphorus reabsorption rate (9/10). None of the children had hypercalciuria. All children were treated with oral phosphorus and calcitriol (8/11 with lack of compliance). Seven children were discharged with a median age of 16.5 years; all had bone deformities, 5/7 had short stature and 1/7 had nephrocalcinosis.
Discussion. Hypophosphataemic rickets is a rare disease with significant long-term morbidity. It should be suspected in children presenting with short stature, developmental delay and orthopaedic abnormalities.
Increased alkaline phosphatase, low serum phosphorus with normal serum calcium and impaired renal tubular reabsorption of phosphate confirm the diagnosis. Early diagnosis and treatment are essential to minimise morbidity in children.
Key-Words: Children; hereditary hypophosphataemic; rickets.
INTRODUCTION
Rickets is a disease of children and adolescents, caused by a failure of growing bone to mineralise.
When this happens in adults after growth-plates fusion, it is called osteomalacia1. Rickets is not a disease only of the past, nor is it limited to developing countries2. Although nutritional rickets from vitamin D deficiency is the most common type of this disease, there are several other forms, unrelated to diet or sunlight exposure1,3 (Table I). In the United States, between 1986 and 2000 less than one-third of children with rickets had nutritional deficiencies, with genetic factors or underlying diseases remaining as the main factors responsible for the illness2. Nowadays the incidence of rickets is not known with accuracy, since there is no national surveillance.
Table I
Types of Rickets
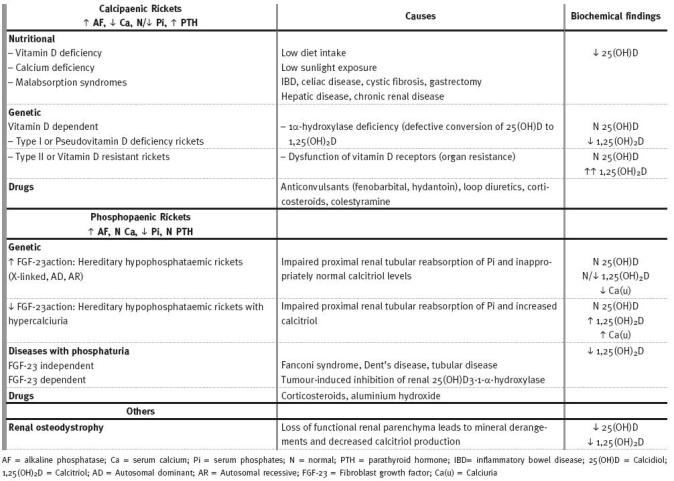
Rickets should be suspected in children presenting with failure to thrive, developmental delay and orthopaedic abnormalities (with asymmetry, pain, or progression in severity). Skeletal changes occur because of the lack of calcified osteoid and the build-up of unossified cartilage1. These are confirmed by characteristic radiographic signs (Table II)2-6.
Table II
Characteristic Skeletal and Radiographic Findings of Rickets

Rickets can be classified into two major groups: calcipaenic and phosphopaenic rickets. While the skeletal findings are similar for both, the extraskeletal manifestations of rickets vary depending upon the primary mineral deficiency (hypoplasia of the dental enamel is typical of calcipaenic rickets, whereas dental abscesses are frequent in phosphopaenic rickets). Serum phosphorus usually is low in phosphopaenic rickets while serum calcium is normal, and may be either decreased or normal in calcipaenic rickets, along with typically elevated parathyroid hormone (PTH). Serum concentrations of 25-OH vitamin D (calcidiol) are low in vitamin D deficiency.
Most of the hypophosphataemic disorders are inherited2,3, including X-linked (XLH), autosomal dominant (ADHR) and autosomal recessive rickets (ARHR) and hypophosphataemic rickets with hypercalciuria.
Hereditary hypophosphataemic rickets is a rare form of disease, with the most common X-linked.
It results from a reduction in the phosphate reabsorption by the renal tubuli, which leads to chronic hyperphosphaturia and hypophosphataemia1,2,6. The tubular maximum reabsorption of phosphate estimates the renal phosphate loss3. There is also a decreased synthesis of calcitriol. Early treatment with phosphate and calcitriol can optimise height outcome and improve the rickets.
Hypophosphataemic rickets with hypercalciuria (HRH) is another rare form of disease, with autosomal recessive heredity. It is distinguished by an increased urinary calcium excretion and elevated plasma calcitriol.
Because HRH is treated with phosphorus supplementation alone, plasma calcitriol levels and urinary calcium excretion must be measured in every patient with phosphopaenic rickets before initiating therapy.
Fibroblast-growth factor 23 (FGF23), a circulating hormone that causes renal phosphate wasting, is increased in XLH rickets, ADHR and ARHR and decreased in HRH.
Tumour-induced osteomalacia (TIO) is an acquired disorder, with many features in common with familial syndromes. It should be considered if the onset of phosphate wasting presents later in childhood or adolescence.
This study intended to characterise the population of children with hypophosphataemic rickets observed over the last 31 years at our unit.
PATIENTS AND METHODS
A retrospective observational study of all children with hypophosphataemic rickets was performed at the paediatric nephrology unit of a tertiary paediatric hospital. Children selected were observed at this unit in a period of 31 years from the year 1982 to 2012.
Patients were identified from the units database, including IT and paper registries. Data collected included demographics, height at birth, height and year of first consultation; diet content in calcium and vitamin D, sunlight exposure, pre-existing medical conditions, previous medications; skeletal, extraskeletal and radiographic findings; laboratory results (alkaline phosphatase, serum calcium and phosphorus, parathyroid hormone, calcidiol (25-OH vitamin D), renal phosphorus reabsorption rate) at diagnosis and evolution; treatment and morbidities; age at discharge. Alkaline phosphatase values were all determined at the same laboratory and normal range was adjusted for childrens age and sex. Stature was evaluated according to 2000 CDC Growth Charts for the United States.
RESULTS
There were 11 children with hypophosphataemic rickets (Table III). Six were diagnosed in the last 12 years and nine were girls. The median age at admission was 4.25 years, varying from ten months to 10.9 years, and the follow-up time had a median of 8.33 years (2.67 to 18.08 years).
Table III
Hypophosphataemic Rickets in the Paediatric Nephrology Unit
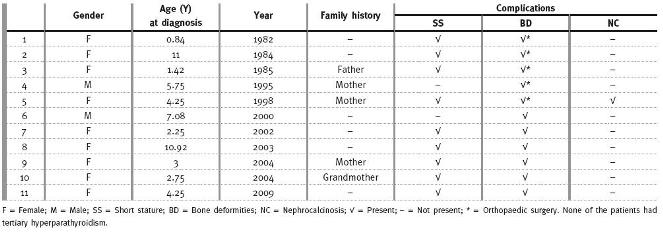
Low sunlight exposure was reported in only one child and lack of vitamin D supplementation in two.
Malabsorption disease and pharmacological habits were not found. Family history of rickets or orthopaedic abnormalities was found in five children (four in a first-degree relative).
The first clinical manifestations were delayed/abnormal walk (seven children) and short stature, present in four children. Of these, one had failure to thrive and another had delayed dental eruption.
Skeletal deformities were present in all children: genu valgum or varum (11/11), thickening of the wrists (7/11), rachitic rosary (4/11), frontal bossing of skull (2/11), Harrisons groove (1/11). Dental abscess was reported in one child and joint pain in six.
At admission all childrens height was below the 5th centile (adjusted for age and gender). Laboratory findings included increased alkaline phosphatase (11/11), median of 400 UI/L (192-665); low serum phosphorus (11/11), with a median of 0.8 mmol/L (0.59-1); serum calcium within the normal range (10/11), median of 2.23 mmol/L (1.8-2.25); normal parathyroid hormone values in six children, increased in five; low renal phosphorus reabsorption rate (9/10), median of 72% (50-83); calcidiol levels available were within the normal range (4/4) and none of the children had hypercalciuria.
The biochemical characteristics were collected for every year of follow-up. Phosphate levels were always low. Calciuria was determined from 24-hours urine at the initial study and posterior values were obtained by calcium/creatinine ratio. Only three children had elevated values, all with lack of compliance: two had hypercalciuria at the last observation and emigrated with their families; one girl had transient elevated calciuria and is now controlled. At the beginning of the study there were few results of PTH, which were available with regular basis only from 1992 forth.
The large majority of these values were elevated.
All children were treated with four or five daily doses of oral phosphorus and calcitriol. Calcitriol dose varied from 10 to 20 ng/kg/dose, twice a day.
Phosphorus was prescribed to achieve 40 mg/kg/day, divided in four or five doses.
Therapy was adjusted to minimise gastrointestinal side effects (diarrhoea) and adjust serum phosphate levels, calciuria and parathyroid hormone. Lack of compliance or irregular medication was reported in eight children, in all cases associated with phosphorus administration.
Seven children, with a median age of 16.5 years, were discharged and transferred to adult medical care. Most of them had short stature: final stature below the 3rd centile (5/7), in the 5th centile (1/7) and in the 10th centile (1/7). Bone deformities of legs persisted in all children and 5/7 were submitted to at least one orthopaedic surgery correction during follow-up. All children were performing renal ultrasonography and there was one case of nephrocalcinosis.
The girl with nephrocalcinosis (stage I bilateral renal echogenicity) was followed from 4 to 17 years old. She did not have hypercalciuria but PTH values were higher than other children (range from 80 to 394 pg/mL). Calcitriol dose was similar. None had tertiary hyperparathyroidism. In three cases the diagnosis of hypophosphataemic X-linked rickets was confirmed by genetic studies.
DISCUSSION
Hypophosphataemic rickets (HR) is a rare genetic disease, due to increased action of FGF23, a hormonelike substance secreted by the osteoblasts, active in renal tubuli, that promotes hypophosphataemia by decreasing the renal phosphorus reabsorption1,3.
Recent experiments suggest that primary action of FGF23 is on vitamin D metabolism, and the effects on phosphate could be secondary (independent of PTH).
FGF23 acts on renal phosphate transport indirectly by influencing vitamin D metabolism and suppressing serum levels of 1,25-vitamin D3 (due to a suppression of the anabolic 25-hydroxyvitamin D3-1(alpha)-hydroxylase (1 α-hydroxylase) and an increase in expression of the catabolic 24-hydroxylase)3,7-11.
PHEX (Phosphate-regulating gene with Homologies to Endopeptidases on the X-chromosome) also plays a major role in renal phosphate handling. It is expressed predominantly in bones and teeth (osteoblasts and odontoblasts) but not expressed in the kidney, which suggests the secondary involvement of a circulating systemic factor. Loss of PHEX function indirectly results in the secretion of specific factors by the osteoblast (phosphatonins, that inhibit renal phosphate handling, and minhibins that inhibit mineralisation). Although physiological substrate for
PHEX remains elusive, peptides of FGF23 and MEPE (Matrix Extracellular Phosphoglycoprotein, a protein that inhibits phosphate-uptake and mineralisation) were proposed8.
The pathogenesis of hypophosphataemic rickets is not fully understood. Increased levels of FGF23 appear to be an important common pathway for hereditary hypophosphataemic rickets and TIO, but the mechanisms for the increased FGF23 vary among these disorders. In XLH rickets, the primary defect is a defective Znmetalloendopeptidase called PHEX, resulting in increased full-length FGF23 and MEPE expression. This causes defects in mineralisation, leading to rickets, dental abnormalities, and/or osteomalacia. The other clinical manifestations of XLH rickets (such as enthesiopathy and dental abnormalities) may also be mediated by mechanisms other than FGF23. In ADHR, activating mutations in FGF23 gene result in proteins resistant to protease cleavage by PHEX or other proteases, and so circulating levels of FGF23 are increased, with consequently inhibition of renal phosphate reabsorption8,9. In TIO, the tumours (typically benign, small and of mesenchymal origin) are the responsible for the production of the phosphaturic hormone (FGF23)8. ARHR is caused by inactivating mutations in the DMP1 gene, which encodes Dentin matrix protein 1 and results in increased FGF23 expression and defective osteocyte maturation.
In this study there were only 11 children over 31 years of study, reflecting the rarity of the disease.
In literature, there was no gender predominance (mostly X-linked dominant hereditarily)12-14 but we report a female predominance (9/11).
The medical history of any child with rickets should include infants gestational age, a detailed dietary history (calcium and vitamin D), the amount of sunlight exposure, medical conditions (malabsorption syndromes, renal disease, malignancy) and medications associated with rickets (loop diuretics, corticosteroids, anticonvulsants and antacids)2,4,5.
Children with HR are usually born with normal length. Since there is an adequate growth velocity in the first years of life, the first clinical manifestations result from the period of slow-growth velocity before diagnosis is made1,2. Skeletal abnormalities appear later in life, with weight bearing after starting to walk, and are less pronounced than in vitamin D deficiency rickets15,16. Family history may allow an earlier diagnosis, around the 6th month of life. In this study the median age at admission was 4.25 years and all children had skeletal abnormalities. The youngest childs mother had diagnosed disease, and the oldest had been followed in several units for short stature and had no family history.
When there is no family history, the diagnosis of rickets in children with compatible clinical and laboratory findings should exclude other diseases that also cause rickets, hypophosphataemia and reduced renal phosphorus reabsorption rate (Hereditary hypophosphataemic rickets with hypercalciuria, Fanconi syndrome, Dents disease, malabsorption syndromes, malignancy).
The evaluation of a child with clinical signs of rickets should include measurement of serum creatinine and liver enzymes to exclude renal insufficiency as the primary aetiology or liver disease as the cause of elevated serum alkaline phosphatase activity. Serum phosphorus and PTH measurements allow the diagnosis of rickets and determine the initial classification of the disease.
Laboratory investigation may include alkaline phosphatase17, serum levels of calcium (total and ionised with serum albumin), phosphorus, parathyroid hormone, urea nitrogen, creatinine, calcidiol and levels of urinary calcium and phosphorus. Serum levels of 1,25 OH2 vitamin D are not stable and should not be used to diagnose vitamin D deficiency or insufficiency.
The characteristic findings in HR are increased alkaline phosphatase, hypophosphataemia, decreased renal phosphorus reabsorption rate and a normal or increased parathyroid hormone, with the remaining values within the normal range. However, laboratory values can vary depending on the stage of the disease and previous medication, which explains the different results in our study.
Genetic studies are the most recent tool to diagnose rickets (the majority of patients with XLH rickets are caused by PHEX mutations; singular cases have been reported with mutations in FGF23, DMP1 and ENPP1)9. Although very helpful confirming the disease, they are expensive and not easily available worldwide (three of our cases were confirmed to be hypophosphataemic X-linked rickets, due to mutations on PHEX gene (Xp22.1)12,14.
The measurement of serum FGF23 levels is a promising laboratory examination18-20. In chronic kidney disease, phosphate levels rise with declining glomerular filtration rate and increase FGF23 levels, to promote renal phosphate excretion; pretreatment circulating FGF23 levels may allow us to predict the refractoriness to calcitriol therapy and increased FGF23 levels are associated with increasing risk of mortality in dialysis patients. In oncogenic osteomalacia, FGF23 is elevated in preoperative serum of patients and normalises after resection of the causative tumor. FGF23 could be also appropriated for the first screening step in determining the etiology of FGF23-related hypophosphataemic rickets. As such, FGF23 appears to be a novel marker in the workup of chronic kidney disease, tumour-induced osteomalacia, and rare genetic causes of rickets.
HR is treated with oral phosphorus and calcitriol. Serum levels of calcidiol should be determined in order to exclude a severe vitamin D deficiency. The main aims of the treatment are to improve growth velocity and avoid or reduce skeletal abnormalities21.
All patients must be carefully monitored after treatment initiation. Radiographic changes may appear within a week, and physical examination findings may normalise within six months.
The earliest biochemical change after treatment initiation is a rise in the level of phosphorus within the first week2. Alkaline phosphatase levels will decrease along the years, achieving normal or just above normal range values2,22.
Adjustments to medications are made to accommodate abnormal fluctuations in serum or urine values: elevated alkaline phosphatase and decreased serum phosphorus require an increase in phosphorus dose21; increased parathyroid hormone requires an increase in calcitriol and decrease in phosphorus.
Calcium to creatinine ratio in spot urine should be determined to detect hypercalciuria2, which require a decrease in calcitriol dose.
Lack of compliance, usually associated with phosphorus multiple administrations, is a common problem in this disease (reported in 8/11 children in our study)23. Along with late diagnosis, this is responsible for the long-term skeletal abnormalities and short stature observed in these patients (all children with bone deformities; 5/7 submitted to at least one surgery and 5/7 children with final stature below the 3rd centile).
Other complications of rickets are nephrocalcinosis (only one child in our study) which justifies performing a renal ultrasonography at 2-5 year intervals. Nephrocalcinosis may occur independently of hypercalcaemia or hypercalciuria, but related to treatment dosages.
Another complication is hyperparathyroidism which may progress to tertiary hyperparathyroidism24.
Growth hormone has been used as adjunctive therapy in HR, but the results are variable25-27.
Increases in serum phosphate and linear growth have been reported, as well as worsening leg deformities, radiographic rickets and no effects on final adult height. There is a lack of clear evidence of benefits of this expensive therapy. It is not approved for this disease in our country.
New therapies are also been investigated. In mouse models, anti-FGF23 completely reversed the hypophosphataemia and associated bone disease. Phase 1 and phase 2 trials of humanised monoclonal anti-FGF23 therapy (KRN23) in humans with X-linked hypophosphataemic rickets are currently underway (A Repeated Study of KRN23 in Adults With X-Linked Hypophosphataemia; principal investigator: Thomas O Carpenter MD, Yale University; KRN23 is an antibody or a mixture of antibodies against FGF23). In conclusion, HR is a rare disease with significant long-term morbidities. It should be suspected in children presenting with failure to thrive, developmental delay and orthopaedic abnormalities. Family history should record any hereditary disease. Infants of affected parents must be screened regularly for hypophosphataemia and increased levels of serum alkaline phosphatase to ensure early treatment and avoid morbidities. Treatment requires monitoring and dose adjustment to prevent complications.
References
1. Filho HM, Castro LC, Damiani D. Hypophosphatemic rickets and osteomalacia. Arq Bras Endocrinol Metab 2006;50:802-3 [ Links ]
2. Nield LS, Mahajan P, Joshi A, et al. Rickets: not a disease of the past. Am Fam Physician 2006;74:619-26 [ Links ]
3. Pettifor JM. Whats new in hypophosphataemic rickets? Eur J Pediatr 2008;167:493-9 [ Links ]
4. Saborio P, Chan JCM. Raquitismo hipofosfatémico renal. In: Nieto G, Santos F, ed. Nefrologia Pediátrica V. Madrid: Aula Médica Ediciones 2000:91-98 [ Links ]
5. Greenbaum LA. Rickets and hypervitaminosis D. In: Kliegman RM, Behrman RE, Jenson HB, Stanton BF, ed. Nelson Textbook of Pediatrics. Philadelphia: Saunders Elsevier, 2007:253-62 [ Links ]
6. Oduwole AO, Giwa OS, Arogundade RA. Relationship between rickets and incomplete distal renal tubular acidosis in children. Italian J Pediatrics 2010;36:54-61 [ Links ]
7. Gattineni J, Baum M. Regulation of phosphate transport by fibroblast growth factor 23 (FGF23): implications for disorders of phosphate metabolism. Pediatr Nephrol 2010;25:591-601 [ Links ]
8. Rowe PS. The wrickkened pathways of FGF23, MEPE and PHEX. Crit Rev Oral Biol Med 2004;15:264-81 [ Links ]
9. Lorenz-Depiereux B, Schnabel D, Tiosano D, et al. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet 2010;86:267-72 [ Links ]
10. Alon US. Fibroblast growth factor (FGF)23: a new hormone. Eur J Pediatr 2011;170:545-54 [ Links ]
11. Liu S, Quarles LD. How fibroblast growth factor 23 works. J Am Soc Nephrol 2007;18:1637-47 [ Links ]
12. Nunes AB, Lazaretti-Castro M. Raquitismo hipofosfatêmico: da clínica à genética molecular. Arq Bras Endocrinol Metab 2000;44:125-32 [ Links ]
13. Mejia-Gaviria N, Gil-Peña H, Coto E, et al. Genetic and clinical peculiarities in a new family with hereditary hypophosphatemic rickets with hypercalciuria: a case report. Orphanet J Rare Dis 2010;5:1 [ Links ]
14. Jap TS, Chiu CY, Niu DM, et al. Three novel mutations in the PHEX gene in Chinese subjects with hypophosphatemic rickets extends genotypic variability. Calcif Tissue Int 2011;88:370-7 [ Links ]
15. Souza MA, Junior LAVS, Santos MA, et al. Dental abnormalities and oral health in patients with hypophosphatemic rickets. Clinics 2010;65:1023-6 [ Links ]
16. Petje G, Meizer R, Radler C, et al. Deformity correction in children with hereditary hypophosphatemic rickets. Clin Orthop Relat Res 2008;466:3078-85 [ Links ]
17. Turan S, Topcu B Gökçe B, et al. Serum alkaline phosphatase levels in healthy children and valuation of alkaline phosphatase z-scores in different types of rickets. J Clin Res Ped Endo 2011;3:7-11 [ Links ]
18. Nelson AE, Bligh RC, Mirams M, et al. Fibroblast growth factor 23: a new clinical marker for oncogenic osteomalacia. J Clin Endocrinol Metab 2003;88:4088-94 [ Links ]
19. Kazama J, Sato F, Omori K, et al. Pretreatment serum FGF-23 levels predict the efficacy of calcitriol therapy in dialysis patients. Kidney Int 2005;67:1120-5 [ Links ]
20. Sridevi Devaraj S, Duncan-Staley C, Jialal I. Evaluation of a method for fibroblast growth factor-23: a novel biomarker of adverse outcomes in patients with renal disease. Metab Syndr Relat D 2010;8:477-82 [ Links ]
21. Filho HCM, Correa PHS. Raquitismo hipofosfatêmico ligado ao X. 1st ed. Brasil: Projeto Diretrizes – Sociedade Brasileira de Endocrinologia e Metabologia, 2004 [ Links ]
22. Mäkitie O, Doria A, Kooh SW, et al. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab 2003;88:3591-7 [ Links ]
23. Garg RK, Tandon N. Hypophosphatemic rickets: easy to diagnose, difficult to treat. Indian J Pediatr 1999;66:849-57 [ Links ]
24. Alon US, Monzavi R, Lilien M, et al. Hypertension in hypophosphatemic rickets: role of secondary hyperparathyroidism. Pediatr Nephrol 2003;18:155-8 [ Links ]
25. Mäkitie O, Kooh SW, Sochett E. Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Clin Endocrinol 2003;58:163-8 [ Links ]
26. Baroncelli GI, Bertelloni S, Ceccarelli C, et al. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphataemic rickets. J Pediatr 2001;138:236-43 [ Links ]
27. Darendeliler F, Bas F, Karaaslan N, et al. The effect of growth hormone treatment on biochemical indices in hypophosphatemic rickets. Horm Res 2001;55:191-5 [ Links ]
Dr Joana S. Caetano
Unidade de Nefrologia Pediátrica
Serviço de Pediatria do Ambulatório
Hospital Pediátrico Carmona da Mota
Avenida Afonso Romão,
3000-602 Coimbra, Portugal
E-mail: sc.joana@gmail.com
Conflict of interest statement.None declared.
Received for publication: 14/09/2012
Accepted in revised form: 16/11/2012

