










Services on Demand
Journal

Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
 Cited by SciELO
Cited by SciELO Related links
 Similars in
SciELO
Similars in
SciELO Share

 Permalink
PermalinkPortuguese Journal of Nephrology & Hypertension
Print version ISSN 0872-0169
Port J Nephrol Hypert vol.33 no.2 Lisboa June 2019
https://doi.org/10.32932/pjnh.2019.07.018
ORIGINAL ARTICLE
Kidney dysfunction in renal amyloidosis: does the complement system play a part in hereditary ATTRV30M and iatrogenic ATTR amyloidosis?
Ana Carina Ferreira MD1,2, Fernanda Carvalho MD1, Ana Rita Santos Dr.1, Fernando Nolasco MD PhD1,2
1 Hospital Curry Cabral, Centro Hospitalar de Lisboa Central (CHLC), Lisbon, Portugal
2 Nova Medical School, Faculdade de Ciências Médicas, UNL, Lisbon, Portugal
ABSTRACT
Introduction and aims: The complement system may play a part in the pathogenesis of amyloid disorders. The primary aim of the study was to analyse the relationship between transthyretin renal amyloidosis (ATTR) and the complement system. The secondary aim was to find potential clinical and morphologic features in ATTR amyloidosis associated with complement activation.
Methods: We performed a retrospective cohort observational study in patients with renal amyloidosis submitted to kidney biopsy and complement measurements from January 2005 to June 2016. We compared clinical and laboratory results in different types of amyloidosis using the Kruskal-Wallis or Fisher’s exact test. We performed a subgroup analysis in ATTR amyloidosis patients, comparing patients with normal and low complement serum levels in terms of clinical, laboratory, and morphologic characteristics, using the Mann-Whitney test or Fisher’s exact test.
Results: We included 42 patients in the analysis: 15 patients had ATTR amyloid deposits, 16 AA amyloid deposits and 11 AL amyloid deposits (all AL λ). A total of ten patients presented low C3 (6 ATTR; 3 AL; 1 AA). None of the patients had low C4 serum levels. Of the 15 Caucasian patients with renal ATTR amyloidosis, 6 (40%) presented low C3. Those patients had a higher sclerotic glomeruli number and 5 out of 6 had C3 deposits in immunofluorescence.
Discussion: In our study, 40% of patients with renal ATTR amyloidosis showed complement consumption or activation, which led us to suspect that ATTR deposits can lead to systemic activation of the alternative complement pathway. It remains to be elucidated if this activation injures peripheral nerves and the kidney.
Conclusions: C3 determinations can prove to be a useful tool for predicting renal involvement and glomerulosclerosis in TTR amyloidosis patients.
Key-words: Amyloidosis, complement system, renal biopsy, transthyretin
INTRODUCTION
Amyloidosis belongs to a group of rare diseases where a particular protein suffers a pathogenic misfolding event in its conformation, with subsequent aggregation and development of insoluble fibril of 8 to 10nm in diameter1. These fibrils acquire a beta-sheet conformation, which produces a green birefringence under polarized light when stained with Congo red, and accumulates in extracellular space of organs leading to organ failure. Currently, 36 different proteins are known to be amyloidogenic and to cause systemic or localized disease in humans2,3. Without treatment, this disorder is usually fatal.
Different amyloid fibrils proteins are known to deposit in renal extracellular space, in glomeruli and/or parenchyma. The classification of the disease depends on the amyloid fibril protein. AL/AH amyloidosis is secondary to accumulation of monoclonal immunoglobulin light or heavy chains1,4; AA amyloidosis is secondary to accumulation of serum amyloid A protein5, and hereditary amyloidosis is secondary to genetic changes in some proteins [such as transthyretin (ATTR), fibrinogen Aα, apolipoprotein AI and AII, and lysozyme]6. Leukocyte chemotactic factor 2 (LECT2), a neutrophil chemotactic factor, which is also a nephropathic form of systemic amyloidosis6-8, has recently been described mainly in Mexican American individuals.
The mechanisms by which amyloidosis leads to kidney dysfunction remain to be elucidated1. Some authors consider that amyloid deposits can cause disruption of tissue architecture, but no study has linked the quantity of amyloid deposits to tissue dysfunction3,9,10. Additionally, amyloid deposits can be toxic to cell function1, especially amyloid protein oligomers or protofibrils3, but how renal damage actually occurs is not known.
In Portugal, most hereditary amyloidosis is due to TTR mutations6. Several studies have shown that mitochondria and complement polymorphisms can modulate penetrance and age of onset of this disease11,12. Renal involvement in ATTRV30M seems to be universal, independent of albuminuria6,9. Nevertheless, kidney dysfunction or degree of proteinuria appears to be related to heavy amyloid deposition in glomeruli and vessels, and is independent of age, duration of the disease and does not parallel with neuropathy.
The complement system, traditionally considered a component of innate immunity, has been implicated in the pathogenesis of several different renal diseases13-16. Moreover, it has been postulated that complement activation plays a role in the pathogenesis of some amyloid disorders, such as Alzheimer’s disease17-19 and even in the pathogenesis of axonal degeneration in hereditary amyloidosis20,21. Given that, we hypothesize that renal dysfunction in ATTR amyloidosis can be mediated by activation of the complement cascade, via focal inflammation, giving rise to tubulointerstitial fibrosis.
The primary aim of the study was to analyse the relationship between ATTR renal amyloidosis and the complement system. The secondary aims were to find potential clinical and morphologic features in ATTR amyloidosis associated with complement activation.
METHODS
We performed a retrospective cohort observational study, including all patients followed at our institution from January 2005 to June 2016, with renal deposits of amyloid confirmed by a kidney biopsy with C3 and C4 measurements at the time of the biopsy. We excluded from the analysis renal transplanted patients at the time of the biopsy.
Predictor variable
The predictor variable was the type of renal amyloidosis, characterized as AL, AA or hereditary ATTR amyloidosis.
Outcome variable
The primary outcome was consumption of complement through C3 or C4 serum levels at the time of renal biopsy, categorized as a binary variable (according to normal or low serum levels).
Subgroup analysis
We performed a subgroup analysis in ATTR patients aiming at finding a link between complement activation and loss of renal function, as well as morphologic features in the biopsy. For this purpose, the secondary outcomes were: 1) renal survival (as a binary variable, defined according to the presence or absence of renal replacement therapy) at the end of follow-up; 2) histomorphologic analysis, by comparing glomerular sclerosis; quantity of amyloid deposits in glomeruli [graded as mild (less than 25%), moderate (25% to 75%) and severe (>75%)]22, in the tubulointerstitium and in vessels [graded as mild (less than 25%), moderate (25% to 50%) and severe (>50%)]22; interstitial fibrosis and tubular atrophy (graded as mild, moderate and severe), vessel lesions (intimal and hyalinosis), and immunofluorescence (IF) findings.
Covariates
Variables analysed were age, gender, race, motive for the biopsy, estimated glomerular filtration rate (eGFR) by EPI formula at the time of the biopsy and at the end of follow-up; serum albumin, 24h proteinuria, and presence of hematuria at the time of renal biopsy. We established follow-up time from the renal biopsy to death, dialysis or renal transplant or, in the absence of the three, to June 2016.
Renal morphology
We reviewed all kidney biopsies stained with hematoxylin-eosin, Masson’s trichrome and silver, assessed the total number and the number of sclerotic glomeruli (excluding the subcapsular glomeruli) and looked for spicules in the basement membrane of glomeruli.
Degrees of interstitial infiltrate, interstitial fibrosis and tubular atrophy were considered mild if the involved cortical was <30%, moderate if between 30 to 60%, and severe if >60%. We evaluated vessel intima status and any hyalinosis. Congo red birefringence detected serum amyloid deposits. We graded amyloid deposits (mild, moderate and severe) in glomeruli, in tubulointerstitial space, and in vessels, and we assessed its localization. The type of amyloid was characterized using IF for light chain kappa and lambda deposits (in case of AL) and also AA. ATTR-type amyloid was detected in embedded paraffin tissue with peroxidase method. Polyclonal antibody rabbit anti-human TTR (A0002, Dako, Denmark) was applied (1/200) for 45 minutes at room temperature. Novolink Polymer kit (RE7140-CE, Leica, Germany) was used according to the manufacturer’s instructions. IF was performed on frozen sections using labeled human immunoglobulin (IgA, IgG, IgM, C3, C4, C1q and fibrinogen). When no frozen fragment was available, we made indirect immunoperoxidase using formalin-fixed paraffin embedded section. The deposits were graded by IF from 0 to 3+.
Statistical analysis
We presented continuous variables as median (IQ interval) and categorical variables as frequencies. The Kruskal-Wallis test or Fisher’s exact test were used to compare patients with the different renal amyloidosis, and their clinical and laboratory results. In the subgroup analysis, the Mann-Whitney test / Wilcoxon ranksum test and Fisher’s exact test were used to compare clinical and laboratory characteristics of patients with normal and low complement serum levels. The evaluation of eGFR at time of diagnosis and end of follow-up was assessed by a Wilcoxon matched pairs signed-rank test.
After the clinical analysis, we performed the morphologic analysis to find potential morphologic features in ATTR amyloidosis associated with complement activation. All data were analysed using STATA package version 13.1. A twosided p<0.05 was considered significance for the whole analysis.
RESULTS
Between January 2005 and June 2016, our renal morphologic laboratory performed 2557 renal biopsies. From these, 43 patients were diagnosed with renal amyloidosis and have serum C3 and C4 measurements (Figure 1): 16 patients had ATTR amyloid deposits, 16 had AA amyloid deposits and 11 had AL amyloid deposits (all AL λ).
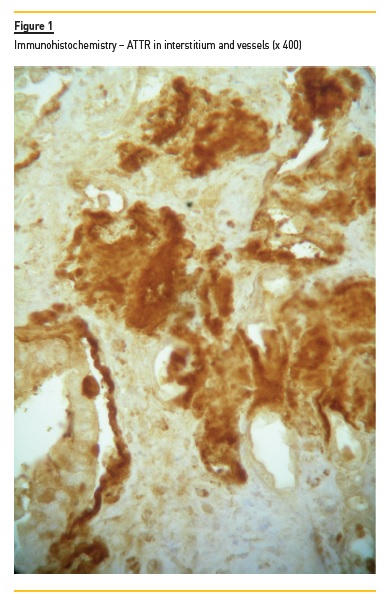
We excluded from the study a young liver-transplanted male with hereditary ATTR amyloidosis, in whom renal biopsy revealed a crescentic post-infectious glomerulonephritis along with ATTR renal amyloidosis.
C3 consumption in the different type of renal amyloidosis
We analysed data from 42 patients (Table 1). At diagnosis, median age was 60 years (51–70), with increasing age from ATTR amyloidosis to AA amyloidosis to AL amyloidosis (p=0.016). Mean eGFR at baseline and at the end of follow-up showed no differences between the different types of amyloidosis. Median 24h proteinuria was 5 (2.9-7.5) g, mean albumin 2.5 (1.9–3.3) g/dl, and patients with AA amyloidosis were the ones with the highest values of proteinuria and lowest values of albumin (Kruskal-Wallis test p=0.006; p<0.001, respectively). There were no reports of malnourished patients.
At the time of the renal biopsy, a total of ten patients presented with low C3 serum levels: 6 out of 15 in ATTR; 3 out of 11 in AL; 1 out of 16 in AA. Although more patients in the ATTR protein group presented with low C3, this finding was not of statistical significance (p=0.05). No patient had low C4 serum levels. We found no differences between patients with low and normal C3 serum levels in the levels of albumin, 24h proteinuria and presence / absence of hematuria.
The median follow-up time was 11 months (2–34), with no differences between the amyloid types. At the end of follow-up, 18 patients were on dialysis (n=9 AA; n=5 ATTR; n=4 AL, p=0.36). When looking at the depletion of C3 serum levels, we found that 60% of patients in the low C3 group were on dialysis at the end of follow-up, although with no statistical significance (p=0.1).
Regarding morphologic features, we found no relevant disparities between renal amyloidosis types, besides the fact that, as a whole, spicules were rare, though frequent in the AL amyloidosis group, where almost 64% of patients presented this finding (p=0.05) and that deposition of amyloid in the medulla was frequent in ATTR patients (46.6%; p=0.0046).
Subgroup analysis – hereditary ATTRV30M and iatrogenic ATTR renal amyloidosis
As stated, we included 15 Caucasian patients (5 males and 10 females) with the diagnosis of renal ATTR amyloidosis, confirmed by a kidney biopsy (Figure 1). One patient with TTR renal amyloidosis along with crescentic post-infectious glomerulonephritis was excluded, as explained.
It should be noted that 9 patients were already diagnosed with hereditary ATTRV30M at the time of the kidney biopsy; in 3 patients hereditary ATTRV30M diagnosis was possible after the kidney biopsy, and in 3 patients iatrogenic ATTR renal amyloidosis was acquired after domino liver transplantation (Table 2).
At the diagnosis, median age was 56 (41–59) years old, median eGFR was 40 (32–85) ml/min, median 24h proteinuria was 4 (1–5) g, and median albumin was 3.7 (3.3–4.1) g/dl; 5 patients presented microhematuria (33.3%). After a median follow-up time of 12 (3–16) months, the eGFR declined to 38 (12.3–57) ml/min, 5 patients started dialysis, 8 patients were liver transplanted (including a double kidneyliver transplant) and 3 patients died (1 male on dialysis and immediately after liver transplantation; 1 female with sepsis, 3 months after liver transplantation, and 1 female with progressive disease).
C3 consumption in patients with ATTR deposits
C3 complement consumption was present in 6 patients (40%), one of these had iatrogenic ATTR amyloidosis. Although he was transplanted 6 years prior due to hepatic failure, his liver function was normal after transplantation and by the time of the renal biopsy. After dividing the ATTR renal amyloidosis patients into two groups according to C3 serum levels (Table 3), we observed that patients with low C3 had a higher sclerotic glomeruli number and a that the quantity of amyloid deposits in the glomerular arterioles was higher compared to those with normal C3 levels (median glomerulosclerosis 3.5 vs. 0; 30.5% vs. 4.7%, p=0.03).
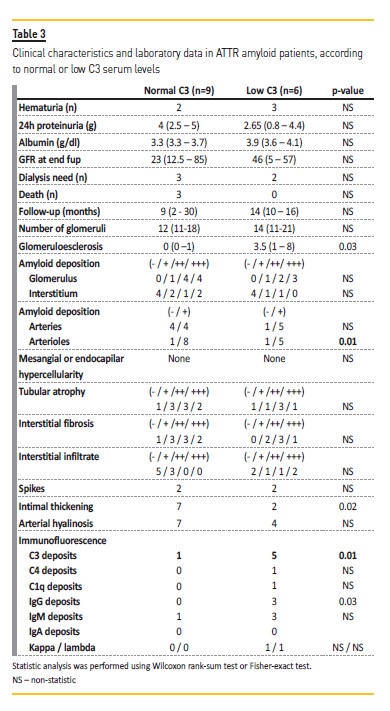
No differences were found in quantity of amyloid deposits in glomeruli, tubules or in interstitial space. Vessel intimal-medial thickening was also more evident in patients with normal C3 levels. Concerning IF deposits, 5 out of 6 patients had C3 deposits on glomeruli (vs. 1/9 of those with normal C3 levels; p=0.01), and 3 out of 6 had IgG deposits as well (vs. none out of 9 patients; p=0.03). Deposits were mainly mesangial, more rarely in parietal walls. The localization of the deposits had no impact on the C3 serum levels.
There was no difference in the final eGFR between the groups, even after applying a survival analysis.
DISCUSSION
Renal amyloidosis is a rare disease which can lead to renal dysfunction by non-clarified means. Complement activation is a well-known event in many kidney disorders, specifically in many glomerulonephritis.
In amyloidosis, this event is not reported. In this retrospective analysis, we studied the potential effect of complement activation on morphologic features and the loss of renal function in patients with renal amyloidosis. We observed three findings: low C3 serum levels are frequent in ATTR renal amyloidosis, but we found no statistical significance when comparing with other types of renal amyloidosis; decreased C3 serum levels associated with C3 deposits in glomeruli (by IF) in ATTR amyloidosis, and decreased C3 serum levels associated with glomerulosclerosis in ATTR amyloidosis.
We must acknowledge the potential limitation in this study. The retrospective and observational nature of a cohort study design with a small sample size prevents us from inferring causality and limits our conclusions; as such this is only a descriptive report. Even more, to know the exact role of complement we need a control group of patients with amyloidosis without renal involvement. Nevertheless, the aim of the study was to see if the involvement of complement cascade in ATTR renal amyloidosis was more frequent than other types of renal amyloidosis.
Even with fifteen patients with ATTR amyloidosis, we were able to show that 40% of those patients had complement consumption or activation through low C3 serum levels. It has been suggested that complement deregulation occurs in neuropathic disorders.
Studies have demonstrated that peripheral nerves synthesize complement factors and regulators and that excessive deposition of complement factors can lead to injury and disease of the peripheral nerve21. Also, Hafer-Macko and co-workers20 demonstrated the presence of complement products around and in the amyloid deposits in acquired (AL) and hereditary (ATTR) amyloid neuropathies, namely C3d, C1q, C4, and C5b-9. Because no Factor Bb was detected, they concluded that the alternative pathway activation was not significant, as it was the classical pathway, in the contribution to the injury of axons in the vicinity of acquired and hereditary amyloid deposits20. Although important, this was a biopsy-based study and no serum levels of complement were measured to understand which complement pathways were involved and the systemic consequences of this local activation. Based on our findings, normal C4 levels and low C3 serum levels, we suspect that ATTR deposits can lead to systemic activation of the alternative complement pathway. It is questionable if this activation injures peripheral nerves and the kidney. Moreover, it should be noted that severity of nephropathy does not correlate with the degree of neuropathy6 and that renal deposition of amyloid does not run parallel with loss of myelinated nerve fibers6.
Complement has been implicated in clinical manifestations of ATTR amyloidosis: we learn from Nylander and co-workers23 that genetic variants of complement factors C3 and isotype C4A may interact with the mutant TTR protein, modifying the expression of the disease24.
In addition, C1q has been implicated in the age of onset in ATTR amyloidosis Cypriot patients11. From this, we can infer that ATTR amyloid fibrils can interact with complement compounds andactivate the cascade, via the alternative pathway, in an attempt to promote phagocytosis, and lysis of the fibrils opsonized.
Complement activation and its role are reported in other types of amyloidosis. In Alzheimer’s disease, many studies have tried to link complement activation and consequent inflammation in the pathogenesis and progression of this disease17-19,25. Concerning AA amyloidosis, in 2004, Hutchinson and colleagues performed experiments to understand the possible complement dependence of AA amyloid deposition in tissues26. They concluded that complement activation by either the classical or alternative pathways was not necessary for the experimental induction of systemic AA amyloid in mice. In our study, we also saw that only one AA amyloidosis patients (out of 16) presented with low C3 serum levels, and that complement activation is not a possible predictor of tissue dysfunction in this type of amyloidosis.
It would be interesting to measure serum C3 levels prospectively in patients with ATTR amyloidosis. Nevertheless, those measurements were not performed. We can speculate that in hereditary ATTRV30M patients already submitted to liver transplantation, C3 can achieve normal serum levels as the amyloid fibrils cease to be produced.
CONCLUSION
To our knowledge, the direct study of the role of complement activation in the progression of renal dysfunction in amyloidosis has not been considered. We believe that, in ATTR amyloidosis, C3 determination can be a useful tool for predicting renal involvement and renal survival. More studies are needed, with more patients, to understand systemic complement activation in ATTR amyloidosis.
References
1. Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17(12):3458-71. [ Links ]
2. Sipe JD, Benson MD, Buxbaum JN, Ikeda SI, Merlini G, Saraiva MJ, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016;23(4):209-13. [ Links ]
3. Picken MM. Proteomics and mass spectrometry in the diagnosis of renal amyloidosis. Clin Kidney J. 2015;8(6):665-72. [ Links ]
4. Dember LM. Modern treatment of amyloidosis: unresolved questions. J Am Soc Nephrol. 2009;20(3):469-72. [ Links ]
5. Obici L, Merlini G. AA amyloidosis: basic knowledge, unmet needs and future treatments. Swiss Med Wkly. 2012;142:w13580. [ Links ]
6. Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol. 2012;7(8):1337- 46. [ Links ]
7. Khalighi MA, Dean Wallace W, Palma-Diaz MF. Amyloid nephropathy. Clin Kidney J. 2014;7(2):97- 106. [ Links ]
8. Murphy CL, Wang S, Kestler D, Larsen C, Benson D, Weiss DT, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis. 2010;56(6):1100-7. [ Links ]
9. Lobato L, Beirao I, Guimaraes SM, Droz D, Guimaraes S, Grunfeld JP, et al. Familial amyloid polyneuropathy type I (Portuguese): distribution and characterization of renal amyloid deposits. Am J Kidney Dis. 1998;31(6):940-6. [ Links ]
10. Snanoudj R, Durrbach A, Gauthier E, Adams D, Samuel D, Ferlicot S, et al. Changes in renal function in patients with familial amyloid polyneuropathy treated with orthotopic liver transplantation. Nephrol Dial Transplant. 2004;19(7):1779-85. [ Links ]
11. Dardiotis E, Koutsou P, Zamba-Papanicolaou E, Vonta I, Hadjivassiliou M, Hadjigeorgiou G, et al. Complement C1Q polymorphisms modulate onset in familial amyloidotic polyneuropathy TTR Val30Met. J Neurol Sci. 2009;284(1-2):158-62. [ Links ]
12. Bonaiti B, Olsson M, Hellman U, Suhr O, Bonaiti-Pellie C, Plante-Bordeneuve V. TTR familial amyloid polyneuropathy: does a mitochondrial polymorphism entirely explain the parent-of-origin difference in penetrance? Eur J Hum Genet. 2010;18(8):948-52.
13. Belgiojoso GB, Tarantino A, Durante A, Guerra L. Complement deposition in glomerular diseases. Proc Eur Dial Transplant Assoc. 1975;11:515-21. [ Links ]
14. Koscielska-Kasprzak K, Bartoszek D, Myszka M, Zabinska M, Klinger M. The complement cascade and renal disease. Arch Immunol Ther Exp (Warsz). 2014;62(1):47-57. [ Links ]
15. Noris M, Remuzzi G. Glomerular diseases dependent on complement activation, including atypical hemolytic uremic syndrome, membranoproliferative glomerulonephritis, and C3 glomerulopathy: core curriculum 2015. Am J Kidney Dis. 2015;66(2):359-75. [ Links ]
16. Popat RJ, Robson MG. Complement and glomerular diseases. Nephron Clin Pract. 2014;128(3-4):238-42. [ Links ]
17. Bergamaschini L, Donarini C, Gobbo G, Parnetti L, Gallai V. Activation of complement and contact system in Alzheimer’s disease. Mech Ageing Dev. 2001;122(16):1971-83.
18. Cribbs DH, Velazquez P, Soreghan B, Glabe CG, Tenner AJ. Complement activation by cross-linked truncated and chimeric full-length beta-amyloid. Neuroreport. 1997;8(16):3457-62. [ Links ]
19. Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ. beta-Amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol. 1994;152(10):5050-9. [ Links ]
20. Hafer-Macko CE, Dyck PJ, Koski CL. Complement activation in acquired and hereditary amyloid neuropathy. J Peripher Nerv Syst. 2000;5(3):131-9. [ Links ]
21. Ramaglia V, Daha MR, Baas F. The complement system in the peripheral nerve: friend or foe? Mol Immunol. 2008;45(15):3865-77. [ Links ]
22. Sen S, Sarsik B. A proposed histopathologic classification, scoring, and grading system for renal amyloidosis: standardization of renal amyloid biopsy report. Arch Pathol Lab Med. 2010;134(4):532-44. [ Links ]
23. Nylander PO, Beckman L, Holmgren G, Steen L. Association of C3 and C4A complement types with familial amyloidotic polyneuropathy. Hum Hered. 1990;40(5):272-7. [ Links ]
24. Ferreira AC, Carvalho F, Nolasco F. Familial amyloidotic polineuropathy and systemic lupus. Lupus. 2012;21(13):1455-8. [ Links ]
25. Reichwald J, Danner S, Wiederhold KH, Staufenbiel M. Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation. 2009;6:35. [ Links ]
26. Hutchinson WL, Herbert J, Botto M, Pepys MB. Classical and alternative pathway complement activation are not required for reactive systemic AA amyloid deposition in mice. Immunology. 2004;112(2):250-4. [ Links ]
Ana Carina Ferreira
Hospital de Curry Cabral, CHLC
Rua da Beneficência nº8 1069-166 Lisbon, Portugal
Email: carina.ferreira@fcm.unl.pt
Disclosure of potential conflicts of interest: none declared.
Received for publication: May 20
Accepted in revised form: May 23


{kind=link}
{kind=link}