











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkINTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic immune complex mediated disease in which disseminated inflammation can affect any organ. Kidney involvement occurs in approximately two thirds of patients1. Lupus nephritis (LN) is a major risk factor for morbidity and mortality and 10% of these patients will develop end‑stage renal disease2.
Nephrotic syndrome is a common presentation of LN, frequently seen in both proliferative (class III, IV) and membranous (class V) LN1.
A rare subset of patients with LN presents with nephrotic syndrome characterized by diffuse foot process effacement (FPE) without immune complex deposition, or only with mesangial immune complex deposition. This entity, lupus podocytopathy, represents approximately 1% of all LN biopsies and remains poorly categorized3‑6,7.
However, the updated 2018 International Society of Nephrology/Renal Pathology Society classification of LN does not include lupus podocytopathy8,9.
CLINICAL CASE
We describe the case of a 49‑year‑old woman with a previous history of SLE. The diagnosis was made when she was 25 years old, with skin, joint and kidney involvement (class IV LN), treated with methylprednisolone and a total of 9.,5 grams of cyclophosphamide.
She underwent a repeat kidney biopsy 8 years later, due to nephritic syndrome, that showed once again a class IV LN, and was re‑treated with cyclophosphamide (another 4.5 grams, performing a cumulative dose of 14 grams).
She has been under no immunosuppression for 6 years, maintaining only hydroxychloroquine (400 mg id) and a RAAS inhibitor. She also developed coronary artery disease and chronic gastritis. The patient sought medical evaluation due to a four‑month course of arthralgias, followed by progressive edema, anorexia and nausea. Physical examination revealed hypertension (184/82 mmHg) and bilateral peripheral edema. Lab results revealed nephrotic range proteinuria (14.2g/day), hematúria hypoalbuminemia (18 g/liter), hyperlipidemia, and KDIGO stage 3 AKI (maximum serum creatinine of 5.19 mg/dl, 0.67 mg/dl 5 months prior). Ultrasonography was unremarkable. Immunologic workup revealed an antinuclear antibody (ANA) titer of 1:329, high titers of anti‑ribonucleoprotein (anti‑RNP) and anti‑Smith (anti‑Sm).
Anti‑double stranded (anti‑dsDNA) and anticardiolipin were negative. There was no complement consumption. Since renal relapse was highly suspected, treatment was promptly initiated with 3 consecutive daily pulses of 1‑gram methylprednisolone, one single dose of cyclophosphamide (500mg), followed by oral prednisolone according to tapering regimen.
A kidney biopsy was performed, showing on light microscopy 22 glomeruli (Figure 1), one of them with global sclerosis, and five with segmental sclerosis (Figure 2). The remaining glomeruli showed swollen podocytes and segmental mesangial proliferation (Figure 2). Immunofluorescence identified mesangial and granular staining of IgA, C3c, C1q, IgM and IgG. Electron microscopy showed diffuse FPE, with only scant immune deposits confined to the mesangium (Figure 3 and Figure 44).
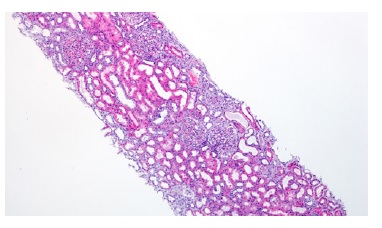
Figure 1: HE 40x. Kidney biopsy showed tubular necrosis, foci of tubular atrophy and interstitial fibrosis.
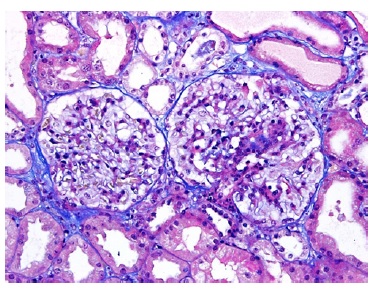
Figure 2: TE 200x. Glomeruli with segmental mesangial proliferation, segmental sclerosis and swollen podocytes.
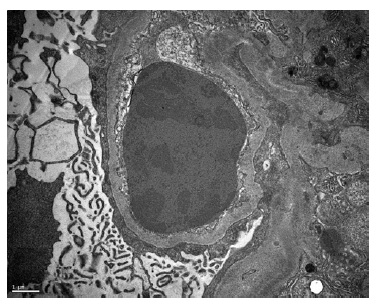
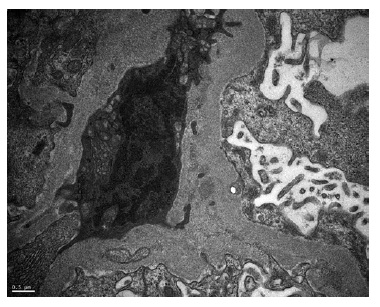
The diagnosis of lupus podocytopathy with focal and segmental glomerulosclerosis (FSGS) pattern was suspected. Treatment was initially resumed to a tappered regimen of oral prednisolone and RAAS inhibitor.
One month later, the patient ́s lab results revealed a serum creatinine of 0.87 mg/dl and proteinuria of 1.5 g/day. Azathioprine was after added in order to steroid sparing and due to the FSGS pattern. Six months after discharge, the patient presented only 300 mg/day of proteinuria.
The severity of the nephrotic syndrome, the diffuse podocyte FPE with scanty immune complex deposition restricted to the mesangial area, and the rapid clinical response to steroids consolidated the diagnosis of lupus podocytopathy.
DISCUSSION
In LN, nephrotic‑range proteinuria generally indicates the presence of a proliferative (class III/IV) or membranous (class V, with or without class III or IV) LN, as a consequence of endocapillary proliferation and imune complex deposition in glomerular capillary walls9. Other causes of nephrotic syndrome should also be excluded, as in the general population.
However, rarely, a patient with nephrotic syndrome and SLE can present with a distinct form of LN.
In 1995, a case of nephrotic syndrome without immune complex deposition within the capillary wall in a SLE patient was reported10.
In 2002, Dube et al. reported 7 patients with SLE and nephrotic syndrome, with kidney biopsy findings of diffuse FPE in the absence of significant peripheral capillary wall immune deposits, consistente with minimal change disease (MCD). In all cases steroid therapy induced a rapid remission4. Hertig et al. reported another 11 patients with SLE and nephrotic syndrome, 4 of them with MCD features and 7 with FSGS. A total of 7 patients were steroid responsive11. The authors observed that the prevalence of idiopathic MCD or idiopathic FSGS was much higher than would be expected by chance, occurring in two per 132 patients with LN at their center.
In 2005, Kraft et al. reported another 8 patients with similar findings. In these series, the frequent correlation of nephrotic syndrome and the onset of SLE led to the suggestion that the podocytopathy is a consequence of active SLE, hence the term lupus podocytopathy5.
In 2016, the largest cohort to date, with 50 cases - 13 with MCD, 28 with MsP and 9 with FSGS - was described by Hu et al7.
Clinical features: Lupus podocytopathy most commonly manifests in females in the third decade, as well as other forms of LN. Nephrotic syndrome is the predominant clinical feature, and frequently the onset symptom of SLE in these patients (88%), and it correlates with SLE activity and extrarenal involvement (most frequently hematological and malar rash)3,7,12. All patients have positive ANAs, but anti‑dsDNA positivity can be as low as 26%. Low C3 is more frequently noted than C4 (68 vs 28%)7.
The FSGS subtype has the highest rate of AKI (78% vs 34% overall)7. Although microscopic hematuria and hypertension are uncommon (18%) in lupus podocytopathy, it is more frequent in the FSGS subtype7,12.
Histopathology and diagnosis: Any of the following three histological patterns can appear on light microscopy. Minimal change disease, MsP and FSGS, and the absence of subepithelial or subendothelial deposition with diffuse FPE (typically >70%) on electron microscopy provides a diagnosis of lupus podocytopathy4,5,7. Mesangial proliferation is the most common histological subtype (56%), followed by MCD (26%) and FSGS (18%), the last being further subclassified as NOS, perihilar, cellular, tip or collapsing lesion7,13. The presence or absence of mesangial proliferation does not seem to significantly change the clinical picture14,15. The severity of proteinuria correlates with the degree of FPE15. The presence of endocapillary hypercellularity, necrosis or crescents, as well as deposits in the subendothelial or subepithelial exclude the diagnosis of lupus podocytopathy. Moreover, tubulointerstitial lesions are more common and severe with the FSGS pattern16. Transition from MCD to FSGS, or to LN class IV or V, on a repeat kidney biopsy has been described.
The proposed criteria for lupus podocytopathy by Hu et al. were: 1) the diagnosis of SLE and nephrotic syndrome 2) compatible light microscopy findings (MCD, MsP, FSGS patterns) 3) immunofluorescence and electron microscopy with deposits which are absent or confined to the mesangium and 4) diffuse podocyte FPE on electron microscopy4‑7,16.
All four criteria should be met and other causes of podocyte injury in SLE should be excluded.
Pathogenesis: Both the absence of deposits in the glomerular capillary wall and the diffuse FPE point to a mechanism that is independente of immune complex deposition in lupus podocytopathy. Podocytes are not only subject to collateral damage due to glomerular capillary lesions secondary immune complexes, but are also a potential direct target in LN17.
This suggests a similar pathogenesis to MCD, in which the production of cytokines and lymphokines by B or T cells, or T‑cell dysfunction, are speculated to cause podocyte injury18, or the presence of potential circulating factor as in primary FSGS19. Also, treatment with interferon has been associated with FSGS and interferons, particularly interferon‑α, are central mediators in the pathogenesis of SLE20,21. In summary, pathogenesis is not yet well understood and needs further research.
Treatment: In general, patients respond well to short courses of high‑dose corticosteroids, as in MCD, but the rate of relapse is very high (89.5%)22. In fact, steroids alone or combined with another immunosuppressive drug induces remission in 94% of cases7. Response and relapse rates differ among the different histological subtypes. FSGS pattern has a disproportionately high rate of non‑response (22% vs 6% overall) and a longer time to achieve remission (median of 8 weeks vs 4 weeks overall) as well as much lower complete remission rates (22% vs 76% overall)7. The subset FSGS with collapsing lesions has even worse outcomes, with end‑stage renal disease in more than 50% of cases, requiring a more aggressive approach in immunosuppression23,24. Patients with apolipoprotein L1 risk variants with SLE and FSGS lesions are even less responsive to therapy25.
More than half of patients have relapses7. Maintenance immunosuppression with an additional drug reduces relapse rates by more than 50%, when compared to corticosteroids alone22. In patients with frequent relapses, a calcineurin inhibitor is suggested because of their stabilizing effects on podocyte injury26.
Prospective studies are needed to confirm the efficacy of diferente treatment regimens.
In our case, some particular aspects deserve consideration. The presence of microscopic hematuria is not frequent in lupus podocytopathy.
Moreover, this was not the inaugural histological diagnosis, as frequently described, and our patient had long‑standing SLE with previously documented LN flares.
However, the histopathological and clinical features are present, and the 4 criteria proposed by Hu et al (above) are all met in our case.
Since FSGS pattern in lupus podocytopathy is less sensitive to glucocorticoid treatment, azathioprine was added in order to improve the remission maintenance, and to reduce steroids’ side effects22.
CONCLUSION
All the above‑mentioned facts argue for lupus podocytopathy to be a distinct entity in LN, and not merely a coexisting histological lesion. It has characteristic clinical‑morphologic features, diferente treatment regimens and prognosis. This emphasizes the relevance of its inclusion in the next revision of LN classification. This case highlights how both nephrologists and pathologists should be aware of this entity when considering the differential diagnosis of a patient with SLE and the nephrotic syndrome.

