










Serviços Personalizados
Journal
Artigo
 Português (pdf)
Português (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Similares em
SciELO
Similares em
SciELO
Compartilhar

 Permalink
PermalinkRevista Portuguesa de Medicina Geral e Familiar
versão impressa ISSN 2182-5173
Rev Port Med Geral Fam vol.28 no.2 Lisboa mar. 2012
RELATOS DE CASOS
Uma causa rara de hipertensão arterial secundária
A rare cause of secondary arterial hypertension
Marta Pessoa,* Carla Sérgio**
*Interna Complementar Medicina Geral e Familiar, USF Buarcos.
**Assistente de Medicina Geral e Familiar, USF Buarcos.
Endereço para correspondência | Dirección para correspondencia | Correspondence
RESUMO
Introdução: A Polineuropatia Amilóide Familiar (PAF) tipo 1, mais conhecida por «Doença dos Pezinhos», é a forma hereditária mais comum de amiloidose, causada pela acumulação de uma proteína mutante (transtirretina). Trata-se duma doença rara e de difícil diagnóstico, principalmente quando não há antecedentes familiares.
Descrição do caso: Mulher de 60 anos de idade, com antecedentes pessoais de hipotiroidismo, dislipidémia, hiperuricémia, perturbação mista ansioso-depressiva, doença venosa dos membros inferiores e Hipertensão arterial (HTA). Em Junho de 2009 foi observada pela primeira vez pela actual médica de família, trazendo resultados de exames laboratoriais que revelavam proteinúria, em sumária de urina. Após pedido de microalbuminúria e cintigrama renal que revelaram insuficiência renal crónica (IRC), foi referenciada para a consulta de Nefrologia do Centro Hospitalar de Coimbra (CHC), onde iniciou estudo etiológico. Em Agosto de 2010 foi internada no Serviço de Nefrologia do CHC com o diagnóstico de insuficiência renal rapidamente progressiva com proteinúria nefrótica. Realizou biópsia renal cujo resultado revelou amiloidose familiar tipo português (ATTR). Foi orientada para a consulta de Paramiloidose do Hospital Geral de Santo António que actualmente frequenta.
Comentário: A discussão deste caso clínico pretende alertar para a importância da realização de uma investigação adequada de um doente com hipertensão arterial. Esta pode revelar factores etiológicos que, mesmo sendo raros e tendo um contexto epidemiológico atípico, devem sempre ser incluídos no processo de raciocínio clínico e diagnóstico diferencial.
Palavras-chave: Polineuropatia Amilóide Familiar; Hipertensão arterial; Nefropatia.
ABSTRACT
Introduction: Familial amyloid polyneuropathy (FAP) type 1, better known in Portugal as “Doença dos Pezinhos”, is a hereditary form of amyloidosis caused by the accumulation of a mutant protein (transthyretin). It is a rare disease which may be difficult to diagnose when there is no previous family history of the condition.
Case description: This report describes the case of a 60-year-old Caucasian woman with a history of hypothyroidism, dyslipidemia, hyperuricemia, mixed anxiety and depressive disorder, chronic venous disease of the legs and hypertension. In June 2009, she was seen for the first time by her new family doctor, with laboratory results that showed proteinuria. A 24-hour urine collection for microalbuminuria and a renal scan were obtained. A diagnosis of chronic renal failure was made. The patient was referred for a nephrology consultation in a central hospital where additional tests were done to determine the cause of her illness. In August 2010, she was hospitalised in the nephrology unit with rapidly progressive renal failure with nephrotic proteinuria. A renal biopsy showed familial amyloidosis, Portuguese type (ATTR). She was then referred to the familial amyloid polyneuropathy clinic in another hospital.
Comment: This case report shows the importance of adequate investigation in a patient with arterial hypertension. It also shows how rare causes may be considered in common clinical syndromes.
Key words: Amyloid Neuropathies; Familial; Hypertension; Kidney Diseases.
INTRODUÇÃO
A HTA é de causa idiopática em mais de 90% dos casos, mas deve-se pensar em causas secundárias perante um utente cumpridor da medicação e das medidas higieno-dietéticas com valores de tensão arterial (TA) de difícil controlo ou com lesão de órgão alvo progressiva apesar do controlo da TA.
Apresenta-se o caso duma mulher com HTA e proteinúria nefrótica rapidamente progressiva, o que fez suspeitar duma causa renal confirmada por biópsia que revelou PAF.
A paramiloidose, amiloidose familiar tipo 1 (português) ou PAF foi descrita pela primeira vez em Portugal, em 1952, por Corino de Andrade.1,2 Sabe-se que, em 2003, existiam mais de 1500 pacientes portadores desta doença em Portugal.3
A PAF tipo 1 é considerada uma doença de transmissão autossómica dominante, endémica em Portugal e de elevada penetrância (mais de 90% dos indivíduos portadores da mutação TTR V30M desenvolvem sintomatologia na idade adulta).1,3,4 O pico de incidência da doença está compreendido entre a terceira e a quarta décadas de vida.1,11 É uma doença progressiva, altamente incapacitante e fatal na maioria dos casos, ocorrendo a morte ao fim de uma média de 11 anos de doença.4-6
Na maioria das situações, os doentes apresentam sintomatologia inespecífica (cansaço fácil e perda de peso), sendo o diagnóstico tardio, quando não há história familiar prévia. Como a própria designação indica, a PAF envolve, mais frequentemente, os nervos periféricos, manifestando-se por neuropatia sensorio-motora e autonómica.3,7-8 No entanto, adopta, na maioria dos casos, proporções de doença sistémica.
Actualmente não existe tratamento médico específico disponível, capaz de inverter o processo de formação de substância amilóide TTR.1 Em termos cirúrgicos, o transplante hepático é considerado o único tratamento eficaz na PAF.1,3,6,8-10 Idealmente, o transplante hepático deverá ser realizado num estádio precoce da doença, ou seja, antes da instalação de qualquer disfunção neurológica e, preferencialmente, no primeiro ano após o início de sintomatologia.1-2,4,7,12
A detecção precoce da doença permite o tratamento atrasando a sua evolução natural, melhorando a sintomatologia, qualidade de vida e sobrevida.
DESCRIÇÃO DO CASO
Identificação e caracterização familiar
M.T.P.R.M., sexo feminino, de 60 anos de idade, raça caucasiana, divorciada, escriturária, natural de Leiria e residente na Figueira da Foz, com o quarto ano de escolaridade.
Vive sozinha, mas tem apoio do filho com quem tem uma relação excelente (Figura 1 e 2). Trata-se duma família unitária, de classe média.

(clique para ampliar ! click to enlarge)
O genograma apresenta-nos os problemas familiares mais relevantes, dos quais se destaca a mãe que faleceu em sequência de IRC de provável etiologia hipertensiva, assim como a história oncológica (neoplasia colo-rectal) presente nas três tias maternas (Figura 1).
Problemas de saúde activos e medicação crónica
• Hipotiroidismo (desde 1998),
• Dislipidémia (desde 2003),
• Perturbação mista ansioso-depressiva (desde 1999),
• Doença venosa dos membros inferiores (desde 2009),
• Hipertensão arterial (desde 2011).
Faz a seguinte medicação crónica: levotiroxina
0,25 mg 1id, losartan 50 mg 2id, amlodipina 5 mg 1id, furosemida 40 mg 3id, sinvastatina 20 mg 1id e alprazolam 0,25 mg 2id.
Hábitos
Alimentação variada e equilibrada, negando hábitos alcoólicos, tabágicos ou consumo de drogas. Não faz actividade física regular. Tem o plano vacinal actualizado e nega ter qualquer tipo de alergias. Episódios esporádicos de insónia inicial controlados com medicação ansiolítica.
Antecedentes Ginecológicos e Obstétricos
Menarca aos 12 anos de idade com ciclos menstruais regulares (cataménios de cinco dias e interlúnios de 28 dias). Histerectomia total com anexectomia subtotal (com remoção do ovário direito) aos 35 anos de idade, por miomatose uterina. Gesta I Para I, gravidez sem intercorrências com parto eutócico, em 1971.
História da doença actual
Consulta 23.06.2009
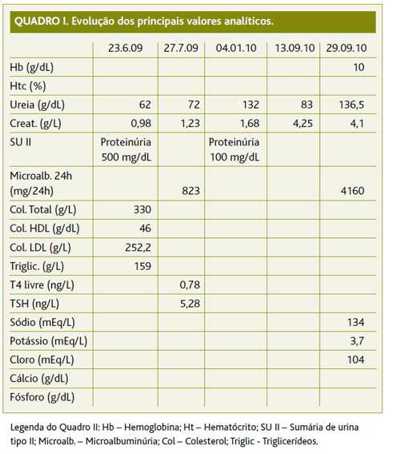
A utente comparece a consulta programada, a primeira com a sua actual médica de família (MF), trazendo resultados de exames laboratoriais e imagiológicos solicitados pela MF anterior. Ao exame objectivo apresenta IMC de 23,4 kg/m2, valor de tensão arterial de 160/80 mmHg, edemas dos membros inferiores, sopro sistólico à auscultação cardíaca, auscultação pulmonar sem alterações. As análises sanguíneas demonstram um valor de ureia elevado, com um valor de creatinina normal, apresentando uma sumária de urina tipo II com proteinúria (Quadro I). A ecografia renal evidenciava «quisto simples no pólo inferior do rim esquerdo com cerca de 4 cm, sem outras alterações».
Foram solicitadas novas análises de sangue e urina com microalbuminúria das 24 horas, electrocardiograma (ECG) e ecocardiograma. Foram aconselhadas medidas higieno-dietéticas com manutenção da medicação já prescrita anteriormente pela sua última médica de família (furosemida 40 mg; losartan com hidroclorotiazida 50 mg/12,5 mg e rosuvastatina 10 mg), assim como vigilância dos valores de tensão arterial em ambulatório.
Consulta 27.07.2009
A utente vem novamente a consulta programada com a sua MF trazendo resultados de exames complementares solicitados. Os valores de tensão arterial em ambulatório mantiveram-se sempre acima de 140/90 mmHg. Ao exame objectivo apresenta tensão arterial de 160/90 mmHg, com diminuição dos edemas dos membros inferiores. As análises demonstram uma elevação do valor da ureia e microalbuminúria de 823 mg/24h (proteinúria nefrótica). As provas de função tiroideia revelaram um hipotiroidismo (Quadro I). O ionograma, níveis de cálcio e fósforo, proteinograma electroforético e ECG não apresentavam alterações.
O ecocardiograma demonstrou «insuficiência mitral e tricúspide muito ligeira». É feito ajuste da medicação hormonal tiroideia, solicitado ecografia da tiróide e cintigrama renal, tendo sido pedida consulta de Nefrologia do CHC. Foi feito reforço das medidas higieno-dietéticas.
Consulta 07.09.2009
A utente apresenta-se em consulta programada com resultados dos exames imagiológicos pedidos na consulta anterior. Ao exame objectivo, o valor de tensão arterial avaliada é de 130/75 mmHg e sem edemas dos membros inferiores. O cintigrama renal revela «rins de dimensões diminuídas com alguma estase intraparenquimatosa; bilateralmente com diminuição da eliminação do fármaco». A ecografia da tiróide demonstra «nódulo hipoecogénico de cerca de 4 mm com área calcificada». Nesta consulta é feito o diagnóstico de IRC sendo solicitadas novas análises sanguíneas enquanto a utente aguardava a consulta de Nefrologia.
Consulta 04.01.2010
Nesta consulta programada, a utente vem informar a sua MF que já foi à consulta de Nefrologia do CHC e que irá iniciar estudo etiológico da IRC. Foi mantida a medicação que já fazia, com aumento da dose da furosemida, alteração do losartan com hidroclorotiazida 50 mg/12,5 mg para losartan 50 mg 2id e amlodipina 5 mg 1id e da rosuvastatina 10 mg para sinvastatina 20 mg id. Foi prescrito ácido fólico, vitaminas do complexo B e calcitriol. Novamente a utente traz consigo resultados das análises solicitadas que demonstram um valor de ureia e creatinina elevados, bem como uma sumária de urina tipo II com proteinúria de 100 mg/dL (Quadro I).
Ao exame objectivo, apresenta uma tensão arterial de 135/80 mmHg. A pedido da consulta hospitalar de Nefrologia, são solicitadas ecografias abdominal, renal e vesical.
Consulta 13.09.2010
A utente comparece, novamente, em consulta programada informando a sua MF que recorreu, no mês anterior, ao Serviço de Urgência (SU) do Hospital Distrital da Figueira da Foz por queixas de disúria e polaquiúria, tendo-lhe sido diagnosticado uma infecção do tracto urinário por Escherichia coli, com agravamento da função renal. A utente é portadora de análises efectuadas neste episódio de urgência. Nesta mesma altura, a utente inicia queixas de naúseas frequentes. Ao exame objectivo, evidenciam-se edemas ligeiros dos membros inferiores e um valor de tensão arterial de 150/85 mmHg. As análises do SU demonstram um agravamento dos valores de função renal sem, no entanto, existirem desequilíbrios hidro-electrolíticos (Quadro I).
São solicitadas novas análises sanguíneas para esta levar à consulta de Nefrologia do CHC.
Consulta 29.09.2010
Cerca de 15 dias depois, a utente vem novamente a uma consulta, trazendo uma carta de alta do Serviço de Nefrologia do CHC, onde esteve internada cerca de 10 dias. A utente recorreu ao Serviço de Urgência daquele hospital por agravamento do quadro de naúseas. Foi-lhe diagnosticada insuficiência renal rapidamente progressiva, tendo realizado biópsia renal, da qual aguarda resultado histológico. Foram adicionados à medicação crónica carbonato de cálcio e eritropoetina.
Os valores analíticos referidos na carta de alta evidenciavam anemia normocrómica normocítica com agravamento dos valores de função renal, urina das 24 horas com 4160 mg de proteinúria e clearance de creatinina de 25,5 ml/min (Quadro I). Realizou estudo da função hepática e tiroideia, serologias víricas e estudo auto-imune, que se revelaram dentro da normalidade.
Ao exame objectivo, foi avaliado um valor de tensão arterial de 145/80 mmHg.
Foi classificada a insuficiência renal crónica em estádio quatro, sendo solicitadas novas análises sanguíneas para a próxima consulta de especialidade.
A utente estava muito ansiosa e apreensiva com o possível resultado da biópsia, pelo que foram esclarecidas todas as dúvidas apresentadas, dando espaço ao diálogo, tendo-se mostrado disponibilidade para a utente regressar ou contactar pelo telefone sempre que necessário.
Consulta 11.10.2010
Em consulta não presencial, são registados os valores das análises solicitadas que revelavam um agravamento da anemia normocrómica normocítica e da função renal, mantendo-se os valores de cálcio e fósforo dentro da normalidade (Quadro I). A utente foi informada destes resultados telefonicamente, tendo sido solicitadas novas análises sanguíneas.
Consulta 04.01.2011
A utente comparece à consulta programada com a sua MF, trazendo consigo o resultado da biópsia renal efectuada durante o internamento em Setembro de 2010. A biópsia renal revela presença de substância amilóide familiar tipo português – TTR positiva. É assim feito o diagnóstico de PAF.
É feita referenciação da utente para a consulta de PAF realizada no Hospital de Santo António, que actualmente frequenta e onde já lhe foi proposto transplante duplo de fígado e rim, para o qual se encontra em lista de espera. O filho da utente foi igualmente chamado a esta consulta específica onde já iniciou investigação complementar.
Nesta consulta, a utente apresenta-se muito ansiosa, apreensiva com o diagnóstico e preocupada com o futuro, nomeadamente com a hipótese do filho poder ter a mesma doença. Foi explicado à utente as principais características da doença, sua monitorização, possíveis tratamentos e prognóstico. Esta, por já ter recolhido muita informação através da internet e por contacto com utentes com PAF, tem uma perspectiva pouco animadora do futuro que se avizinha.
COMENTÁRIO
No caso da utente cujo caso clínico é aqui descrito, a constatação de valores de tensão arterial descontrolados, associados a resultados de função renal alterados e com agravamento progressivo, fizeram colocar a hipótese de se estar perante um caso de hipertensão secundária e o estudo etiológico da hipertensão arterial foi, desde início, dirigido para uma provável causa renal. O valor de microalbuminúria de 823 mg/dL (proteinúria nefrótica) fez desde logo levantar a suspeita (Quadro I) e o cintigrama renal confirmou estar-se em presença de uma IRC. É feita a referenciação da utente para os Cuidados de Saúde Secundários para diagnóstico etiológico.
O agravamento da função renal, despoletado pela infecção do tracto urinário e o início do quadro de naúseas, desencadearam o internamento no Serviço de Nefrologia do CHC. O estudo da função hepática e tiroideia, serologias víricas e estudo autoimune excluiu prováveis causas de IRC. A biópsia renal foi necessária para identificar a provável causa da IRC e o estudo histológico detectou a presença de substância amilóide do tipo português – TTR positiva, fazendo o diagnóstico de PAF TTR positiva.
Existem condições que na literatura científica consultada são apresentadas como pontos-chave da PAF e que, neste caso clínico, se mostram desfavoráveis a este diagnóstico. São exemplo disso a idade do início de sintomatologia, a dúvida sobre a existência de antecedentes familiares, a ausência de queixas demonstrativas de polineuropatia sensório-motora como sintomatologia inicial, assim como de sintomatologia típica de disfunção autonómica (alteração dos hábitos intestinais e do esvaziamento gástrico, hipotensão postural ou queixas genito-urinárias).2
Relativamente à idade de início da sintomatologia, a PAF é uma patologia que se manifesta, com maior frequência, entre a terceira e a quarta décadas de vida. No entanto, existem alguns casos de início tardio da doença descritos na literatura científica.13
Um dos pontos a favor do diagnóstico de PAF num indivíduo é a história familiar. No entanto, em 13% dos casos podem não existir antecedentes familiares.13 Neste caso clínico a utente apenas soube informar que a sua mãe também era hipertensa e que faleceu já a realizar hemodiálise, situação que leva a suspeitar que, provavelmente, esta também sofreria de PAF. O único familiar que actualmente demonstrou interesse em realizar investigação complementar foi o filho da utente, que já foi consultado no Hospital de Santo António. O estudo que realizou foi negativo, não sendo, portanto, portador desta mutação génica e da doença da mãe.
Outro dos pontos favoráveis ao diagnóstico de PAF é a polineuropatia sensorio-motora que não foi motivo de queixa por parte da utente (nomeadamente parestesias dos membros inferiores). No entanto, segundo informações da equipa de enfermagem, a utente recorreu recentemente aos cuidados desta para tratamento de escoriações e lacerações indolores nos pés, o que permite concluir que já poderá haver algum grau de neuropatia amilóide.
A PAF é uma doença que geralmente adquire proporções de doença sistémica, condicionando, com bastante frequência, a qualidade de vida dos doentes. A equipa de saúde (médica e enfermeira de família) tem tido a preocupação de, além de proporcionar os devidos cuidados de saúde, dar o apoio psicológico que esta utente necessita. Apesar de se tratar de uma família unitária, a utente sempre referiu ter grande apoio psicológico, funcional e económico por parte de familiares próximos e amigos. Essa situação foi avaliada e confirmada através de alguns métodos de avaliação familiar (Figura 1 e 2).
O papel do MF perante patologias menos frequentes, como é o caso da PAF, não é de menor importância. Apesar da maioria destes doentes, senão a totalidade deles, ter seguimento em consultas periódicas nos Cuidados de Saúde Secundários e Terciários, é ao MF que cabe acompanhá-los em ambulatório, durante toda a vida. A articulação entre Cuidados de Saúde Primários e Secundários/Terciários é de extrema importância, principalmente nos doentes crónicos, portadores de doença sistémica e incapacitante. É fundamental a monitorização contínua e a prevenção de lesões secundárias de órgãos alvo, devendo o MF receber informação de retorno por parte dos cuidados hospitalares, de forma a também poder participar, de forma activa, nesta vigilância periódica. Igualmente, é também no MF que estes doentes encontram o apoio psicológico e conforto emocional de que tanto necessitam, de modo a se sentirem motivados para enfrentarem, diariamente, as limitações e as complicações impostas pela doença.
Em termos de vigilância periódica, o MF deve ter em atenção quais os principais sistemas/órgãos alvo que a PAF poderá atingir e redobrar a sua vigilância sobre estes (Quadro II):
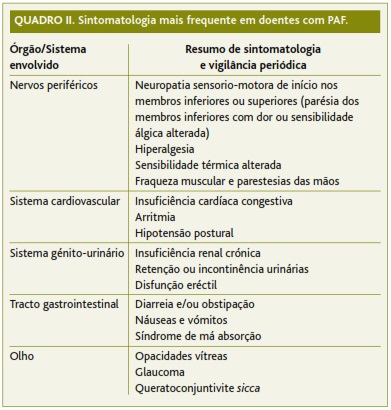
• Sistema cardiovascular: a vigilância deve ser realizada com ecocardiograma.14 Igualmente, poderá ser oportuna a realização periódica de ECG de 12 derivações e/ou cintigrama de perfusão de miocárdio.14
• Sistema neurológico: a monitorização deve incluir um exame neurológico sumário, com especial atenção ao exame do pé, testando os vários tipos de sensibilidade (álgica, vibratória e proprioceptiva).12,14-16 O MF deve igualmente questionar o doente sobre sintomatologia reveladora de disfunção autonómica (alteração dos hábitos intestinais e do esvaziamento gástrico, hipotensão postural, retenção ou incontinência urinárias e disfunção eréctil).2,12
• Olhos: a prevenção primária do aparecimento de opacidades vítreas por deposição de substância amilóide deve ser efectuada através de consulta de Oftalmologia periódica.14
• Sistema renal: a vigilância da função renal deverá incluir monitorização periódica dos valores de creatinina e ureia.8,14 A detecção de microalbuminúria representa um estádio precoce de nefropatia amilóide e geralmente precede a instalação de neuropatia.6-7,17
A instalação de insuficiência renal crónica origina anemia por deficiência de eritropoetina, situação muito frequente nos estádios tardios da doença (presente em 25% dos pacientes com PAF tipo 1 após uma média de 9 anos de doença) e cuja correcção deverá ser feita através da administração da variante recombinante humana desta hormona.5,14,17-20
O transplante hepático ortotópico (THO) é a única opção terapêutica actual com resultados favoráveis para os doentes com PAF. É realizado amplamente como terapia génica cirúrgica (95% da proteína TTR mutante é produzida no fígado).21
Em termos de sintomatologia, o THO demonstra estabilização e alguma regressão da neuropatia autonómica, melhoria da neuropatia periférica e deterioração mais lenta da função renal, relativamente aos doentes que não realizaram THO.6,8
Caso a função renal esteja gravemente deteriorada, como é o caso da utente descrita neste caso clínico, que apresenta uma clearance de creatinina de 25,5 mL/min, deverá ser ponderado o transplante duplo de fígado e rim.20,22-24 Recentemente foi comunicado à MF que a utente iria iniciar hemodiálise, situação que irá manter enquanto aguarda a realização do transplante duplo de fígado e rim para o qual já foi proposta.
Por tudo o que foi exposto, pretende-se, com a descrição deste caso clínico, alertar para a necessidade de ser sempre realizada investigação de causas secundárias de hipertensão arterial em doentes com este diagnóstico de novo, ou naqueles em que esta entidade já é anteriormente conhecida mas que apresentem alterações analíticas e/ou imagiológicas desajustadas à patologia. A etiologia primária ou idiopática da hipertensão arterial é a mais frequente mas as causas secundárias não poderão ser descuradas, podendo estas, ao serem eliminadas, levar ao controlo dos valores tensionais. A suspeita diagnóstica e a investigação dirigida a esta são duas situações em que o MF tem um papel fundamental e privilegiado, tal é a proximidade que tem em relação à comunidade e a continuidade de cuidados que lhe presta.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Roberts J, Oglesby R. Amyloidosis, Transthyretin-Related. Emedicine [Internet]. Wolf R, medical editor. Talavera F, pharmacy editor. Brent L, managing editor. Mechaber A, CME Editor. Besa E, chief editor. 2009 [actualizado em 15/09/2009; acedido em 04/01/2011]. Disponível em http://emedicine.medscape.com/article/335301. [ Links ]
2. Monteiro E, Freire A, Barroso E. Familal amyloid polyneuropathy and liver transplantation. J Hepatol 2004 Aug; 41 (2): 188-194. [ Links ]
3. Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol 2005 Jul; 62 (7): 1057-62. [ Links ]
4. Vaz C, Couto M, Duarte C, Salvador MJ, Inês L, Malcata A. Um caso invulgar de dor generalizada: paramiloidose simulando fibromialgia. Acta Reumatol Port 2009 Abr-Jun; 34 (2): 431-5. [ Links ]
5. Beirão I, Lobato C, Costa PM, Fonseca I, Mendes P, Silva M, et al. Kidney and anemia in familal amyloidosis type I. Kidney Int 2004 Nov; 66 (5): 2004-9. [ Links ]
6. Rocha A, Lobato L, Silva H, Beirão I, Santos J, Pessegueiro H, et al. Characterization of end-stage renal disease after liver transplantation in transthyretin amyloidosis (ATTR V30M). Transplant Proc 2011 Jan-Feb; 43 (1): 189-93. [ Links ]
7. Lobato L, Beirão I, Silva M, Bravo F, Silvestre F, Guimarães S, et al. Familial ATTR amyloidosis: microalbuminuria as a predictor of symptomatic disease and clinical nephropathy. Nephrol Dial Transplant 2003 Mar; 18 (3): 532-8. [ Links ]
8. Carvalho MJ, Lobato L, Ventura A, Beirão I, Rocha G, Seca R, et al. Remission of proteinuria following liver transplantation for familial amyloid polyneuropathy TTR Met30. Transplant Proc 2000 Dec; 32 (8): 2664-6. [ Links ]
9. Dattilo P. Familial (ATTR) amyloidosis misdiagnosed as the primary (AL) variant: a case report. Cases J 2009 Dec 9; 2: 9295. [ Links ]
10. Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation 2004 Jan 15; 77 (1): 64-71. [ Links ]
11. Kolstoe SE, Mangione PP, Belotti V, Taylor GW, Tennent GA, Deroo S, et al. Trapping of palindromic ligands within native transthyretin prevents amyloid formation. Proc Natl Acad Sci U S A 2010 Nov 23; 107 (47): 20483-8. [ Links ]
12. Tomás MT, Santa-Clara H, Monteiro E, Carolino E, Freire A, Barroso E. Alterações da força de preensão em portadores de polineuropatia amiloidótica familiar. Acta Med Port 2010 Set-Out; 23 (5): 803-10. [ Links ]
13. Rudolph T, Kurz MW, Farbu E. Late-onset familial amyloid polyneuropathy (FAP) Val30Met without family history. Clin Med Res 2008 Sep; 6 (2): 80-2. [ Links ]
14. Sekijima Y, Yoshida K, Tokuda T, Ikeda S. Familial Transthyretin Amyloidosis. National Center for Biotechnology Information [Internet]. 2001. [actualizado em 15/09/2009; acedido em 04/01/2011]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK1194. [ Links ]
15. Ohmori H, Ando Y, Makita Y, Onouchi Y, Nakajima T, Saraiva MJ, et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familal amyloidotic polyneuropathy. J Med Genet 2004 Apr; 41 (4): e51. [ Links ]
16. Mendonça T, Pires MM, Reis E, Reis H, Lobato L,Coelho T. Neuropatia periférica como apresentação de amiloidose primária. Sinapse 2005; 5 (2): 90. [ Links ]
17. Lobato L. Familial Amyloidotic Polyneuropathy: how transthyretin associated amuyloidosis involves the kidney. Port J Nephrol Hypertens 2008 Jan-Mar; 22 (1): 23-30. [ Links ]
18. Beirão I, Moreira L, Barandela T, Lobato L, Silva P, Gouveia CM, et al. Erythropoietin production by distal nephron in normal and familial amyloidotic adult human kidneys. Clin Nephrol 2010 Nov; 74 (5): 327-35. [ Links ]
19. Beirão I, Lobato L, Moreira L, Costa PM, Fonseca I, Cabrita A, et al. Long-term treatment of anemia with recombinant human erythropoietin in familial amyloidosis TTR V30M. Amyloid 2008 Sep; 15 (3): 205-9. [ Links ]
20. Beirão I, Almeida S, Swinkels D, Costa PM, Moreira L, Fonseca I, et al. Low serum levels of prohepcidin, but no hepcidin-25, are related to anemia in familial amyloidosis TTR V30M. Blood Cells Mol Dis 2008 Sep-Oct; 41 (2): 175-8. [ Links ]
21. Lanchman H, Hawkins P. Amyloidosis, Familial Renal. Emedicine [Internet]. Feinfeld D, medical editor. Talavera F, pharmacy editor. Aronoff G, managing editor. Schmidt R, CME editor. Batuman V, chief editor. 2009 [actualizado em 29/10/2009; acedido em 04/01/2011]. Disponível em: http://emedicine.medscape.com/article/246221. [ Links ]
22. Adams D, Samuel D, Gloulon-Goeau C, Nakazato M, Costa PM, Feray C et al. The course and prognostic factors of familial amyloid polyneuropathy after liver transplantation. Brain 2000 Jul; 123 (Pt 7): 1495-504. [ Links ]
23. Lobato L, Beirão I, Silva M, Fonseca I, Queirós J, Rocha G, et al. End-stage disease and dialysis in hereditary amyloidosis TTR V30M: presentation, survival and prognostic factors. Amyloid 2004 Mar; 11 (1): 27-37. [ Links ]
24. Lobato L, Ventura A, Beirão I, Miranda HP, Seca R, Henriques AC, et al. End-stage renal disease in familial amyloidosis ATTR Val30Met: a definitive indication to combined liver-kidney transplantation. Transplant Proc 2003 May; 35 (3): 1116-20. [ Links ]
25. Holmes Jr R, Baethge B, Edison J. Amyloidosis, Overview. Emedicine [Internet]. Talavera F, pharmacy editor. Brent L, managing editor. Mechaber A, CME Editor. Diamond H, chief editor. 2009 [actualizado em 30/07/2009; acedido em 04/01/2011]. Disponível em http://emedicine.medscape.com/article/335414. [ Links ]
26. Rakela J. Recurrencia de la enfermedad hepática primaria después del transplante hepático. Rev Med Chile 2010 Abr; 138 (4): 504-10. [ Links ]
27. Conselho Editorial da Revista de Clínica Geral. Normas para apresentação de artigos à Revista Portuguesa de Clínica Geral. Rev Port Clin Geral 2010 Mai-Jun; 26 (3): 325-40. [ Links ]
28. Empis C. Por norma ler as normas. Rev Port Clin Geral 2011 Jan-Fev; 27 (1): 11-2. [ Links ]
29. Murphy J, Lloyd M. Hypertension. In: Hogan MJ, editor. Mayo Clinic Cardiology Concise Textbook. 3rd ed. Rochester, MN: Mayo Clinic Scientific Press; 2007. p. 741-749. [ Links ]
30. Snanoudj R, Durrbach A, Gauthier E, Adams D, Samuel D, Ferlicot S, et al. Changes in renal function in patients with familal amyloid polyneuropathy treated with orthotopic liver transplantation. Nephrol Dial Transplant 2004 Jul; 19 (7): 1779-85. [ Links ]
Endereço para correspondência | Dirección para correspondencia | Correspondence
Marta Cristina da Costa Pessoa
Rua de Santo António, 74
3050-456 Pampilhosa
CONFLITOS DE INTERESSE
As autoras declaram não ter interesses financeiros, ligações pessoais ou outros conflitos de interesse que possam enviesar o presente trabalho.
Recebido em 22/08/2011
Aceite para publicação em 19/03/2012

