










Serviços Personalizados
Journal

Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
 Similares em
SciELO
Similares em
SciELO Compartilhar

 Permalink
PermalinkGE-Portuguese Journal of Gastroenterology
versão impressa ISSN 2341-4545
GE Port J Gastroenterol vol.27 no.3 Lisboa jun. 2020
https://doi.org/10.1159/000503075
CLINICAL CASE STUDY
Malignant Peritoneal Mesothelioma as a Rare Cause of Dyspeptic Complaints and Ascites: A Diagnostic Challenge
Mesotelioma peritoneal maligno como causa rara de queixas dispépticas e ascite: Um desafio diagnóstico
Susana Marques de Sousaa, Filipa Pereirab, Maria Duartea, Marta Marquesa, Dolores Vázqueza, Cristina Marquesa
aInternal Medicine Department, Centro Hospitalar Póvoa de Varzim/Vila do Conde, Póvoa de Varzim, Portugal; bOncology Department, Instituto Português de Oncologia do Porto, Porto, Portugal
* Corresponding author.
ABSTRACT
Introduction: Malignant peritoneal mesothelioma (MPM) is a rare malignancy of the mesothelial cells in the peritoneum. The best-defined risk factor is asbestos exposure, but germline mutations in BAP1 also increase susceptibility to this tumor. The diagnosis of MPM is challenging since clinical manifestations are often nonspecific. Case Presentation: We describe a case of MPM in a 53-year-old former construction worker with prior asbestos exposure. The clinical presentation was a 3-month history of dyspeptic complaints. As initial workup, abdominal ultrasound and upper gastrointestinal endoscopy were performed. Chronic gastritis due to Helicobacter pylori was detected, which was promptly treated but without symptom relief. Abdominal ultrasound showed small volume ascites with hyperechogenic foci, which was later confirmed on computed tomography scan showing the presence of peritoneal nodules in the greater omentum and mesentery. A thorough investigation was conducted based on the suspicion of peritoneal carcinomatosis. A non-peritoneal primary tumor was not found. Ascitic cytology and immunocytochemical studies were suggestive of mesothelioma. He underwent exploratory laparotomy and inoperable peritoneal disease was observed. Peritoneal biopsy confirmed epithelioid-type MPM. Systemic therapy was initiated with platinum plus pemetrexed with good response. The last follow-up was 38 months after the diagnosis. Discussion/ Conclusion: The diagnosis of MPM is challenging since it requires a high degree of suspicion. MPM has a poor prognosis. The standard of treatment recommended is cytoreductive surgery with hyperthermic intraperitoneal chemotherapy. For those who are inoperable, systemic therapy with pemetrexed-cisplatin combination is the alternative. Given the infrequency of disease, it is imperative to ensure patient participation in clinical trials with the purpose of treatment standardization.
Keywords: Mesothelioma, Peritoneal neoplasms, Dyspepsia, Ascites
RESUMO
Introdução: O mesotelioma peritoneal maligno (MPM) é uma neoplasia rara das células mesoteliais no peritoneu. O fator de risco mais estabelecido é a exposição aos asbestos, mas as mutações germinativas BAP1 também aumentam a suscetibilidade a esse tumor. O diagnóstico é desafiador, uma vez que as manifestações clínicas são frequentemente inespecíficas. Caso Clínico: Reporta-se o caso de um homem de 53 anos, ex-trabalhador da construção civil, com história de exposição a asbestos. O doente apresentava um quadro de queixas dispépticas com 3 meses de evolução. Para estudo inicial, foram realizadas ecografia abdominal e endoscopia digestiva alta. Na endoscopia foi documentada gastrite crónica por Helicobacter pylori tendo feito tratamento de erradicação, mas sem melhoria sintomática. A ecografia abdominal revelou ascite de pequeno volume com focos hiperecogénicos em suspensão, o que foi posteriormente confirmado em tomografia computorizada, demonstrando a presença de nódulos peritoneais no grande epíplon e mesentério. Uma investigação exaustiva foi conduzida na suspeita de carcinomatose peritoneal, mas não foi encontrado nenhum tumor primário noutra localização. A citologia do líquido ascítico e os estudos imunocitoquímicos foram sugestivos de mesotelioma. O doente foi submetido a uma laparotomia exploradora na qual se documentou doença peritoneal inoperável. A biópsia peritoneal confirmou MPM do tipo epitelióide. Foi iniciado tratamento de quimioterapia com platino em associação com pemetrexed, com boa resposta. Seguimento atual de 38 meses pós-diagnóstico. Discussão/Conclusão: O diagnóstico de MPM é desafiante uma vez que requer um alto grau de suspeição. MPM tem um prognóstico reservado. O tratamento standard recomendado é a cirurgia citorredutora com a quimioterapia intraperitoneal hipertérmica. No entanto, nos doentes inoperáveis, a alternativa é a quimioterapia sistémica com pemetrexed-cisplatina. Dada a raridade da doença, é imperativo integrar estes doentes em ensaios clínicos com o objetivo de padronização do tratamento.
Palavras-Chave: Mesotelioma, Neoplasias peritoneais, Dispepsia, Ascite
Introduction
Malignant peritoneal mesothelioma (MPM) is a rare malignancy of the mesothelial cells in the peritoneum with an incidence in Europe of 1.2–1.3 cases per million per year [1]. Mesotheliomas can also occur in other serosal membranes, like pleura, pericardium, and tunica vaginalis of the testes. Of all mesotheliomas, pleural mesotheliomas are more frequent than MPM [2]. The link with asbestos exposure is weaker in MPM than in pleural mesothelioma (33–50 vs. > 80%), but it is still the best-defined risk factor [3]. The latency period between the exposure and the diagnosis ranges from 20 to 50 years [2]. MPM usually presents in the fifth and sixth decades of life and is more common in men [4]. In the early stages of the disease, clinical manifestations are nonspecific, such as early satiety, anorexia, weight loss, vomiting, constipation, and/or diarrhea. Ascites and growth of tumor masses result in abdominal distension and abdominal pain. Less frequent presentations include new-onset hernia, fever of unknown origin, and intestinal obstruction or perforation. However, approximately 8% of patients are diagnosed incidentally [5]. A nonspecific clinical presentation may pose a diagnostic challenge for physicians and that is why diagnosis is often delayed and MPM usually detected at advanced stages.
Case Presentation
We report a case of a 53-year-old male, ex-smoker and a former construction worker with a prior 5-year exposure to asbestos beginning at 14 years of age. He had also a first-degree family medical history of MPM – his brother. However, the medical team only had access to this information later in the course of the clinical investigation. The patient was initially evaluated by his primary care physician for a 3-month history of dyspeptic complaints (epigastric abdominal pain, early satiety, nausea, and occasional vomiting). He had no constipation, diarrhea, gastrointestinal blood loss, asthenia, or weight loss. Physical examination was normal. Initial workup showed small volume ascites with hyperechogenic foci on abdominal ultrasound. Upper gastrointestinal endoscopy revealed antral erythematosus gastropathy (gastric biopsy: chronic gastritis with intestinal metaplasia and evidence of Helicobacter pylori microorganisms). Helicobacter pylori eradication treatment was done but did not completely resolve the symptoms.
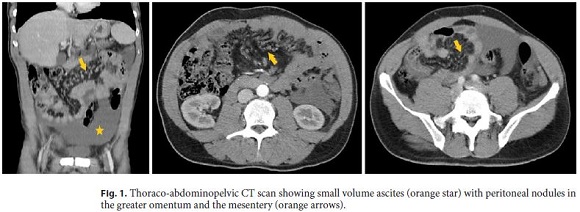
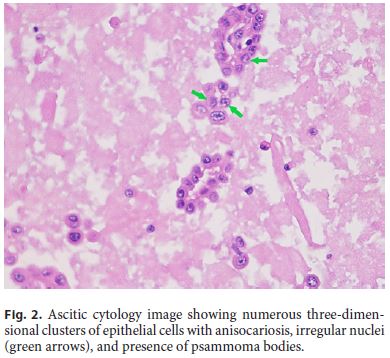
The patient was then referred to the Internal Medicine Department with the diagnosis of suspected peritoneal carcinomatosis. Laboratory values did not show abnormalities. Tumor markers (alpha-fetoprotein, CA125, CA19-9, and carcinoembryonic antigen) were within normal limits. Thoraco-abdominopelvic computed tomography (CT) scan (Fig. 1) exhibited mild centrilobular emphysema with bronchial thickening in the medium pulmonary lobe and small volume ascites with peritoneal nodules in the greater omentum and the mesentery. Bronchofibroscopy, tracheobronchial brush cytology, and bronchoalveolar lavage were normal including negative Ziehl-Neelsen staining. Colonoscopy only showed an adenomatous polyp with low-grade dysplasia in the sigmoid colon. Finally, ultrasound-guided paracentesis was done with the removal of a turbid serous liquid characterized by serumascites albumin gradient of 0.65 g/dL, total protein of 5.8 g/dL, lactate dehydrogenase of 780 U/L, glucose of 27 mg/dL, and normal adenosine deaminase and amylase. Ascitic fluid cell count revealed 695 white blood cells/mm3 with 74% lymphocytes. Gram stain of the fluid was negative. Ascitic cytology (Fig. 2) showed numerous three-dimensional clusters of epithelial cells with anisocariosis, irregular nuclei, and presence of psammoma bodies. These cells expressed calretinin, CD10, and CK7 in the immunocytochemical studies.
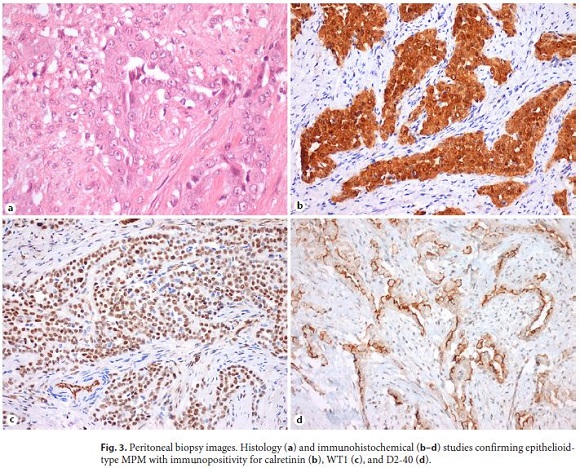
For the suspicion of mesothelioma, the patient was referred to a tertiary care oncology center for further management. Positron emission tomography (PET) scan was done, which revealed high 18F-FDG uptake foci at peri-hepatic, peri-gastric, and peri-splenic sites and anterior abdominal wall. No extra-abdominal dissemination was seen. The patient was evaluated by a multidisciplinary team and he was proposed for cytoreductive surgery plus hyperthermic intraperitoneal chemotherapy (HIPEC). A diffuse intraperitoneal and a bulky disease in parietal peritoneum with liver, stomach, and colon transverse invasion was seen during laparotomy, so the surgery could not be done. Peritoneal biopsy (Fig. 3) confirmed epithelioid-type MPM based on histologic findings and immunohistochemical studies (calretinin+, WT1+, D2-40+, EMA+, AE1/AE3+, vimentin+, CDX2–, focal CK7+, CK20–, focal CEA+).
Chemotherapy with cisplatin and pemetrexed was initiated. In the 5th cycle, the patient had a hypersensitivity reaction grade 2 and cisplatin was suspended, but he maintained pemetrexed in monotherapy. He completed 8 cycles of pemetrexed with stable disease as best response. Three months after the end of treatment, peritoneal disease progression was observed and the patient started carboplatin plus pemetrexed (6 cycles) with partial response. The patient maintains follow-up with stable disease. The last follow-up was 38 months after the diagnosis.
Discussion
MPM is an uncommon tumor, whose diagnosis can be a challenge since clinical manifestations are often nonspecific. The diagnosis is based on medical history along with radiological, laboratory, and histopathological findings. The main imaging modalities used are CT scan, magnetic resonance imaging, and PET scan, although there is no pathognomonic imaging finding for MPM [5]. Usually, the first-line modality is abdominal CT scan with intravenous contrast. On CT scan, MPM appears as a solid, heterogeneous, soft tissue mass with irregular margins that is enhanced by contrast or multiple small nodules involving the serosa. A diffuse distribution throughout the abdominal cavity, with no lymph node involvement or distant metastases, should raise suspicion of MPM since it tends to be more expansive than infiltrative [6]. The MPM differential diagnosis includes peritoneal carcinomatosis, serous peritoneal carcinoma, ovarian carcinoma, lymphomatosis, and tuberculous peritonitis. A peritoneal biopsy is essential for definitive diagnosis of MPM, which should be based on both appropriate histologic morphology and immunohistochemical findings. Histologically, MPM is divided into epithelioid, sarcomatoid, and biphasic subtypes. The epithelioid subtype is the most frequent (75–90%) and is associated with the best prognosis [7]. The optimal immunohistochemical panel for distinguishing MPM remains to be clearly defined. There is a consensus that EMA, calretinin, CK5/6, WT-1, HBME-1, thrombomodulin, podoplanin, mesothelin, and D2-40 are immunoreactive in MPM [8]. More recently, it was found that immunohistochemical loss of nuclear BRCA-1 associated protein 1 (BAP1) is present in almost 50% of pleural mesotheliomas and two-thirds of MPM, but in less than 1% of high-grade serous carcinomas [9]. Moreover, germline mutations in BAP1 increase susceptibility to various cancer types. The most common are pleural and peritoneal mesotheliomas, uveal and cutaneous melanomas, and renal cell carcinomas. Testing for BAP1 mutations should be made in patients with mesothelioma occurring at a young age (< 50 years old) and in patients with multiple family members affected by mesothelioma or other cancers associated with germline BAP1 mutations [10]. In our patient, the search for germline BAP1 mutation was not done because he had only one family member (his brother) with the diagnosis of MPM and also because his brother had a heavier exposure history to asbestos (> 10 years). Both worked together as construction workers in a time when the use of asbestos was not prohibited. In Portugal, this prohibition only occurred in 2005. Besides, the exposure in our patient begun in adolescence and this may be associated with a greater risk for development of malignant mesothelioma [11]. In our patient, the time latency between the initial exposure to the diagnosis of MPM was 39 years. Thus, both MPM cases were considered to be linked to occupational asbestos exposure and not related to family history and possible germline BAP1 mutation. Besides, there were no other family members with other cancers associated to this mutation.
MPM is a life-threatening malignancy that, with no treatment, has a median overall survival of 8.4 months [12]. The standard of treatment is cytoreductive surgery with HIPEC, which increased the median survival to more than 5 years [3, 13]. For those who are inoperable, systemic therapy is the alternative. Pemetrexed-cisplatin combination has become the new standard of care for patients with unresectable malignant mesothelioma [14]. Two studies showed that pemetrexed in combination with cisplatin for MPM had a disease control rate of 71.2% and the median overall survival was 13.1 months [15, 16]. Replacing cisplatin with carboplatin showed to have a similar efficiency [3]. Besides an inoperable disease, our patient had a good response to systemic therapy with much longer survival so far.
Conclusion
MPM is a rare disease with various diagnostic challenges that require a high degree of suspicion. MPM continues to have a poor prognosis because there are limited therapeutic options. Given the rarity of disease, it is imperative to ensure patient participation in clinical trials with the purpose of treatment standardization.
References
1 Gatta G, van der Zwan JM, Casali PG, Siesling S, Dei Tos AP, Kunkler I, et al.; RARECARE working group. Rare cancers are not so rare: the rare cancer burden in Europe. Eur J Cancer. 2011 Nov;47(17):2493–511.
2 Yang H, Testa JR, Carbone M. Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr Treat Options Oncol. 2008 Jun;9(2-3):147–57.
3 Broeckx G, Pauwels P. Malignant peritoneal mesothelioma: a review. Transl Lung Cancer Res. 2018 Oct;7(5):537–42.
4 Salo SA, Ilonen I, Laaksonen S, Myllärniemi M, Salo JA, Rantanen T. Epidemiology of malignant peritoneal mesothelioma: A population-based study. Cancer Epidemiol. 2017 Dec;51:81–6.
5 Boussios S, Moschetta M, Karathanasi A, Tsiouris AK, Kanellos FS, Tatsi K, et al. Malignant peritoneal mesothelioma: clinical aspects, and therapeutic perspectives. Ann Gastroenterol. 2018 Nov-Dec;31(6):659–69.
6 Kim J, Bhagwandin S, Labow DM. Malignant peritoneal mesothelioma: a review. Ann Transl Med. 2017 Jun;5(11):236. [ Links ]
7 Munkholm-Larsen S, Cao CQ, Yan TD. Malignant peritoneal mesothelioma. World J Gastrointest Surg. 2009 Nov;1(1):38–48.
8 Husain AN, Colby TV, Ordóñez NG, Allen TC, Attanoos RL, Beasley MB, et al.; Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Guidelines for Pathologic Diagnosis of Malignant Mesothelioma 2017 Update of the Consensus Statement From the International Mesothelioma Interest Group. Arch Pathol Lab Med. 2018 Jan;142(1):89–108.
9 Andrici J, Jung J, Sheen A, DUrso L, Sioson L, Pickett J, et al. Loss of BAP1 expression is very rare in peritoneal and gynecologic serous adenocarcinomas and can be useful in the differential diagnosis with abdominal mesothelioma. Hum Pathol. 2016 May;51:9–15.
10 Kittaneh M, Berkelhammer C. Detecting germline BAP1 mutations in patients with peritoneal mesothelioma: benefits to patient and family members. J Transl Med. 2018 Jul;16(1):194. [ Links ]
11 Carbone M, Kanodia S, Chao A, Miller A, Wali A, Weissman D, et al. Consensus Report of the 2015 Weinman International Conference on Mesothelioma. J Thorac Oncol. 2016 Aug;11(8):1246–62.
12 Kaya H, Sezgı C, Tanrıkulu AC, Taylan M, Abakay O, Sen HS, et al. Prognostic factors influencing survival in 35 patients with malignant peritoneal mesothelioma. Neoplasma. 2014;61(4):433–8.
13 Sugarbaker PH. Update on the management of malignant peritoneal mesothelioma. Transl Lung Cancer Res. 2018 Oct;7(5):599–608.
14 Garcia-Carbonero R, Paz-Ares L. Systemic chemotherapy in the management of malignant peritoneal mesothelioma. Eur J Surg Oncol. 2006 Aug;32(6):676–81.
15 Jänne PA, Wozniak AJ, Belani CP, Keohan ML, Ross HJ, Polikoff JA, et al. Open-label study of pemetrexed alone or in combination with cisplatin for the treatment of patients with peritoneal mesothelioma: outcomes of an expanded access program. Clin Lung Cancer. 2005 Jul;7(1):40–6.
16 Carteni G, Manegold C, Garcia GM, Siena S, Zielinski CC, Amadori D, et al. Malignant peritoneal mesothelioma-Results from the International Expanded Access Program using pemetrexed alone or in combination with a platinum agent. Lung Cancer. 2009 May;64(2):211–8.
Statement of Ethics
This study did not require informed consent nor review/approval by the appropriate ethics committee.
Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Sources
There were no funding sources.
* Corresponding author.
Susana Marques de Sousa
Internal Medicine Department
Centro Hospitalar Póvoa de Varzim/Vila do Conde
Largo da Misericórdia, PT–4490-421 Póvoa de Varzim (Portugal)
E-Mail susana.m.sousa.g@gmail.com
Received: July 2, 2019; Accepted after revision: August 31, 2019
Acknowledgement
We thank Dr. Conceição Saldanha from the Department of Cytology at Unilabs (Porto) and Dr. Rui Henrique from the Department of Pathologic Anatomy at Instituto Português de Oncologia do Porto, for providing the cytology and histological images.
Author Contributions
Susana Sousa designed and wrote the article and also participated in acquisition, analysis, and interpretation of data. Filipa Pereira and Cristina Marques were the assistant medical team and critically revised the article. Filipa Pereira also participated in acquisition, analysis, and interpretation of data. Maria Duarte, Marta Marques, and Dolores Vázquez made contributions on the design of the article and acquisition of data and critically revised the article. All the authors approved the final version to be published.

