











 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Similares em
SciELO
Similares em
SciELO 
 Permalink
PermalinkIntroduction
Neuroendocrine neoplasms (NENs) can arise from different organs, even though the lungs, gastrointestinal (GI) tract, and pancreas are the most common sites. GI and pancreatic NENs (panNENs) are histologically classified under the same category; however, panNENs have particular clinical features from GI NENs and, thus, should be considered separately. PanNENs have been historically regarded as rare, but the reported prevalence in autopsy series (0.8-10%) is much higher than in population-based studies and their incidence has raised more than 6-fold over the last 3 decades [1]. This growth occurred across all disease stages and tumour grades, but it was particularly pronounced for localized low-grade tumours due to the widespread use of advanced imaging tests [1].
PanNENs are divided into functional and non-functional tumours. About 70-90% of panNENs are classified as non-functional [2]. Functional tumours secrete particular hor-mones or peptides, such as insulin, gastrin, vasoactive intestinal peptide, glucagon, and somatostatin.
Inherited syndromes are important to recognize in the setting of panNENs, as they have significant implications for patient’s medical management, representing an opportunity for early detection of subsequent manifestations. Although most panNENs are sporadic, approximately 10% are due to an inherited syndrome, most commonly multiple endocrine neoplasia type 1 (MEN1) and, less commonly, von Hippel-Lindau disease (VHL), neurofibromatosis type 1 (NF-1), or tuberous sclerosis complex (TSC) [3]. Mahvash disease, a recently described inherited disease, has also been associated with panNENs.
In this review, the Portuguese Pancreatic Club summarizes the classification, diagnosis, and staging of panNENs, with a focus on imaging studies. It also summarizes the characteristics and particularities of the diagnosis and treatment of panNENs associated with inherited syndromes.
PanNEN Diagnosis
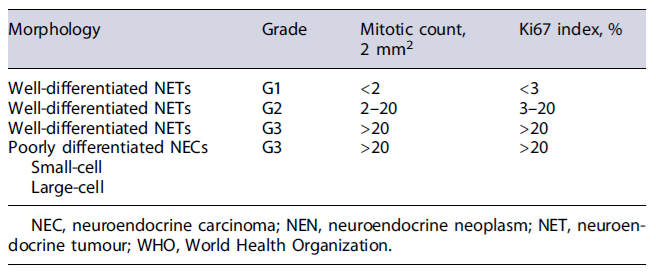
In accordance with World Health Organization (WHO) criteria, panNEN definitive diagnosis requires histological confirmation [4]. For appropriate pathological diagnosis, morphology, grading and immunohistochemical staining for chromogranin A (CgA) and synaptophysin should be reported. The neuroendocrine phenotype is confirmed by the immunohistochemical detection of one of the two neuroendocrine markers commonly used: synaptophysin and CgA. Other neuroendocrine markers, such as neuron-specific enolase and CD56, can be positive in panNEN, but lack specificity [5]. According to WHO 2019 classification, NENs are classified according to morphology and proliferation (Ki67 index and mitotic count) into well-differentiated NENs (G1 to G3) and poorly differentiated neuroendocrine carcinomas (NEC) (always G3) (Table 1) [4]. If Ki67 index and mitotic count are discordant, the higher grade is attributed [6]. Well-differentiated NENs and NEC are biologically and genetically two different diseases. Furthermore, clear prognostic differences can be seen between these two conditions, even in grade 3 NEN. Therefore, the most recent WHO classification split the heterogeneous G3 NEN into well-differentiated NEN G3 and poorly differentiated NEC G3 [4, 6].
Clinical suspicion of functional panNENs can be confirmed by the measurement of specific hormones secreted by functional tumours (i.e., insulin, proinsulin, glucagon, gastrin, vasoactive intestinal polypeptide) and its correlation with hormonal symptoms [5]. Hormone levels also correspond to changes in tumour burden and can therefore serve as specific tumour markers during follow-up. If insulinoma is suspected, serum level of insulin and C-peptide along with glucose during prolonged fasting (72 h) is useful for the diagnosis [7]. Patients with insulinoma present abnormally elevated levels of insulin and C-peptide during hypoglycemia. Proinsulin levels are also elevated in insulinoma. When gastrinoma is suspected, fasting serum gastrin levels should be evaluated. A fasting serum gastrin level that is 10 times greater than the upper limit of the normal range along with a gastric pH <2 is diagnostic of gastrinoma [8]. Demonstration of gastric acid hypersecretion by means of gastric fluid pH evaluation in particular can be obtained through nasogastric tube or upper endoscopy using electrode, filter paper, or biochemical evaluation of aspirate [9]. However, given the advent of highly sensitive imaging-techniques and problems with the classical diagnosis of gastrinomas (intermediate levels of fasting serum gastrin and widespread use of proton pump inhibitors) most expert centres have adopted an alternative diagnostic work-up that include upper endoscopy, endoscopic ultrasonography, magnetic resonance imaging (MRI), and somatostatin receptor (STTR) imaging [9]. Serologic CgA has a limited specificity for the diagnosis of panNEN; however, it might be useful in follow-up [10]. Immunohistochemistry staining for peptide hormones such as gastrin, insulin and glucagon can also help confirm the source of a clinical symptomatology, but there is no complete agreement between immunohistochemistry and symptomatology [11]. Approximately 10% of non-functional panNENs are multiple (multifocal) [10].
PanNEN Staging
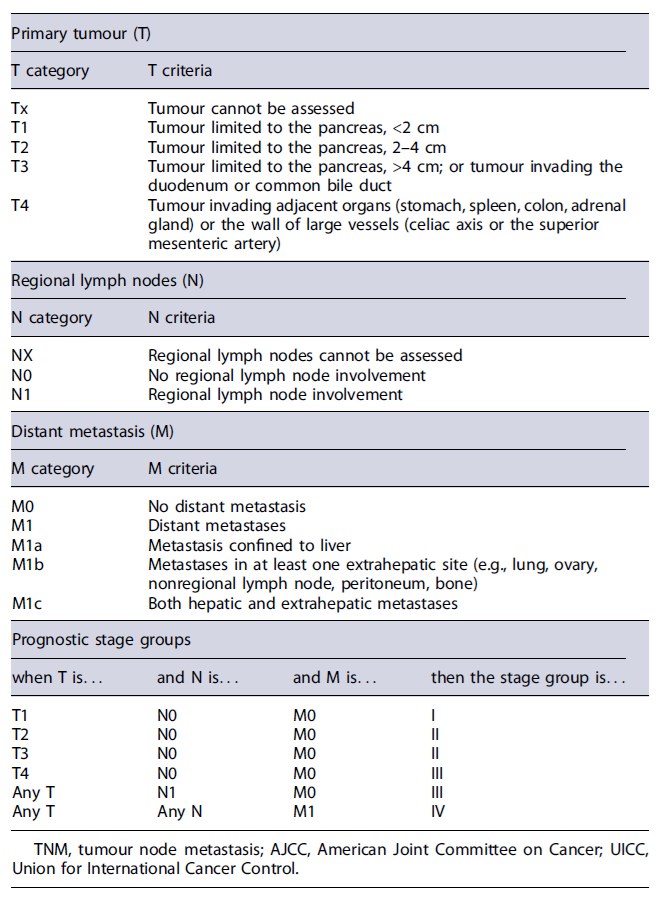
Disease stage and tumour grade are the two major independent prognostic parameters and should always be assessed to select the best therapeutic strategy. Tumour size >2 cm and Ki67 index >3% are predictors of metastatic disease, which is associated to decreased survival. Regarding panNENs staging, the tumour, node, and metastasis staging system proposed by the European Neuroendocrine Tumour Society (ENETS) and adopted in the eighth edition of the American Joint Committee on Cancer (AJCC) is recommended (Table 2) [12]. For NECs, however, the staging system of adenocarcinomas should be applied [12]. Furthermore, the primary tumour site has also an impact on the prognosis in advanced disease and should be detected in the presence of metastatic NEN. In fact, data show that patients with metastatic panNENs have a less favourable prognosis than patients with metastatic small intestinal NENs [13].
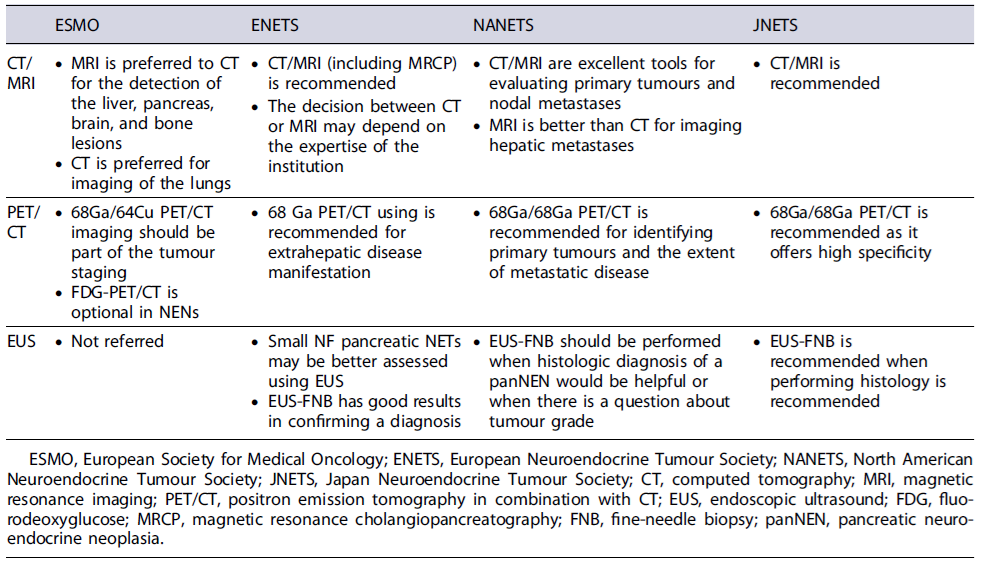
Computed tomography (CT) is the most widely used method for NEN imaging because of its availability and high diagnostic yield [14]. The ENETS, North American Neuroendocrine Tumour Society (NANETS), and European Society for Medical Oncology (ESMO) consensus guidelines recommend pancreatic protocol CT (three-phase CT) as the best imaging modality for staging, primarily due to the characterization of vascular involvement of the primary tumour. The sensitivity of CT to detect panNENs is 61-93% and the specificity is 71-100% [14-16]. The detection rate for liver metastases (LMs) is 79% [17, 18], and for extra-abdominal soft tissue metastases is 70% [19]. However, despite the good diagnostic performance of CT, this radiological method shows lower than desired sensitivity for the detection of small metastatic lymph nodes (<1 cm), bone metastases, and small peritoneal metastases [20].
MRI is a relevant complementary imaging test in the staging of patients with panNENs. In fact, abdomino pelvic MRI protocol is advantage ousfor examination of the liver and the pancreas, when compared with CT, and is also recommended in the initial staging, as it outperforms CT for imaging small LMs [11]. Currently, diffusion-weighted imaging with MRI as well as magnetic resonance cholangiopancreatography is routinely applied, facilitating lesion detection. The MRI sensitivity to detect panNENs is described as 54-100%. The sensitivity of MRI for detection of LMs is 82-98%[21]. MRI is also superior to CT for imaging of the bones and the brain. However, MRI may miss small lung metastases, and CT is preferred for imaging of the lungs [14]. Despite this, MRI protocols, similar to CT protocols, are usually restricted to images from the 11th vertebra body through the iliac crest in the staging of panNENs.
Endoscopic ultrasound (EUS) is the current optimal imaging method to diagnose small panNENs, being able to detect lesions as small as 2-3mmindiameter.EU Sisreported as having 82-93% sensitivity and 86-95% specificity for the detection of panNENs [22]. When compared with CT, EUS appears to have a better diagnostic performance, being able to detect lesions not apparent in other diagnostic modalities [23]. In the ENETS consensus guidelines, EUS is the imaging study of choice when other non-invasive imaging studies are negative, allowing screening the entire pancreas and a detailed evaluation of the tumour [5]. EUS has been shown to be superior to CT in the detection and localization of panNENs in patients with MEN1 syndrome, where the tumours are usually small and multifocal [24]. Furthermore, contrast-enhanced EUS (CE-EUS) is helpful in characterizing small panNENs, which are incidentally found on other imaging modalities [25]. Over 90% of panNENs showed hypoechogenicity in B-mode and hyperenhancement after the injection of contrast agent in contrast-enhanced EUS and up to 75% of hypervascular lesions on CE-EUS were NENs in a recent study [26]. Another benefit of EUS is that it allows for tissue acquisition, using fine-needle aspiration for cytology or, fine-needle biopsy (FNB) with a cutting needle for histopathological diagnosis. Several studies have documented better diagnostic performance with end-cutting FNB needles, particularly for Ki67 index determination (tumour grading), which may be underestimated in fine-needle aspiration samples [27, 28]. Preoperative knowledge of tumour grading is relevant for treatment decision, particularly in small (<2cm) panNENs, for which tumour grading should be considered in the choice between surgery and surveillance [10, 26].
When diagnosing most NENs, STTRs imaging with positron emission tomography in combination with CT (PET/CT) using radiolabelled somatostatin analogues (e.g.,[68Ga]DOTATOC, [68Ga]DOTANOC, [68Ga]DOTA-TATE) is highly sensitive and should be included in the tumour staging process [14]. It offers a high detection rate of lymph node, bone, and peritoneal lesions, as well as a high detection rate of primary lesions in patients with unknown primary tumours [11]. In the past, when PET/CT was not as accessible, STTR scintigraphy (OctreoScan™)wasused, with significantly less sensitivity [11]. The sensitivity to detect panNEN by STTR-PET/CT is 70.5-87.0%, and the specificity is 75-100% [29]. For the detection of bone metastases, STTR-PET/CT shows a sensitivity of 97-100%and a specificity of 92-100% [30]. Notwithstanding, the potential for false-positive uptakes must be considered, particularly within the uncinate process and the pancreatic head [28]. STTR-PET/CT should also not be used in the differential diagnosis between panNENs and other hyper-vascular nodules, such as ectopic spleen - for characterizing pancreatic nodules detected on MRI or CT, EUS with bi-opsy is superior [10].
[18F]FDG-PET/CT is an additional diagnostic tool for patients with panNENs, particularly those equal to or higher than G2, characterized by higher glucose metabolism and lower STTR expression than low-grade NENs [31]. Furthermore, it provides prognostic value since FDG-positive NEN lesions associate with a worse prognosis [31, 32]. Combined STTR and FDG-PET/CT imaging (dual tracer PET/CT) have shown complementary lesion detection [31]. However, the benefits of this combination are not validated and should only be adopted on an individual basis, balancing the potential advantages with the increasing costs [11]. Regarding diagnosis and staging of NECs, FDG-PET/CT is of central importance, since STTR-PET/CT has low sensitivity.
Other diagnostic techniques such as contrast-enhanced ultrasound (US) and intraoperative US might be useful in the localization and staging of pan-NENs [11]. In fact, intraoperative US has an excellent diagnostic performance for the detection of lesions located in the pancreas and liver and is mandatory before pancreatic resection in MEN1 syndrome patients [11].
In summary, disease staging is a major independent prognostic parameter and should always be assessed. CT or MRI protocols with abdominopelvic imaging should be performed with additional anatomical segments evaluation in case of findings suggestive of metastatic disease. Based on current evidence, MRI should be preferred for the detection of the liver, pancreas, brain, and bone lesions, while CT is preferred for imaging of the lungs. Whole-body STTR-PET/CT imaging is complementary to CT or MRI and should also be part of the tumour staging. A summary of the imaging staging recommendations from different international societies can be found in Table 3.
Genetic Syndromes Associated with panNENs and Particularities of Their Clinical Approach
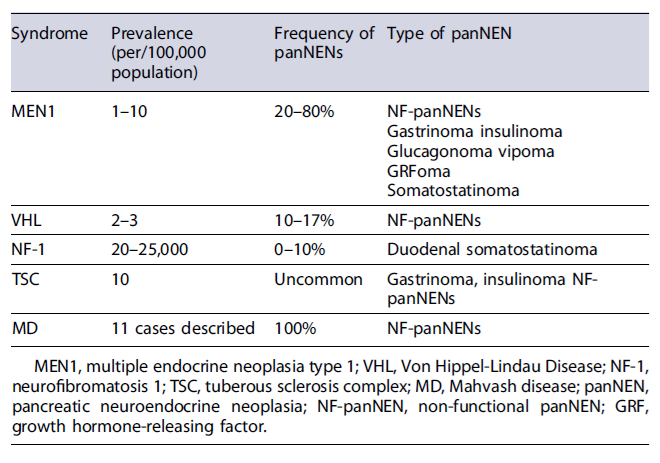
Five different inherited syndromes are associated with the development of panNENs: MEN1, VHL, NF-1, also known as Von Recklinghausen disease, TSC (Table 4), and the more recently described Mahvash disease. Mahvash disease (the rarest among the five known hereditary panNEN syndromes) is the only recessively in-herited PNET syndrome (associated with a mutation in the glucagon receptor gene), has a penetrance of 100%and appears to be exclusively associated with the development of non-functional panNENs. So far, only 11 cases have been describe in the literature, however, the clinical impact of Mahvash disease on panNENs is likely higher than that of NF-1 and TSC, which have very low panNEN penetrance [33]. The first four hereditary panNEN syndromes will be further discussed below.
Multiple Endocrine Neoplasia Type 1
MEN1 is a rare, autosomal-dominant, syndrome secondary to mutations in the MEN1 gene on chromo-some 11q13, which encodes the protein menin. Menin has an important role in regulating cell growth, cell cycle progression, and various other cellular processes [34]. Classically, MEN1 is characterized by the development of tumours/hyperplasia in multiple endocrine tissues (parathyroid, pancreas, and pituitary); however, other tumours can also be associated with MEN1, including the adrenal, skin, thyroid, CNS, smooth muscles as well as carcinoid (lung and thymus) tumours and gastric NENs [34-38].
As much as 20-80% of all patients with MEN1 will develop clinically relevant panNENs (Table 4) [34]. MEN1 occurs in 20-30% of all patients with gastrinomas and Zollinger-Ellison syndrome (ZES), 5% of patients with insulinomas, and <3% with non-functional pan-NENs [34]. In other types of functional panNENs, known as rare functioning tumours, MEN1 is the most frequent familial condition associated, with glucagonomas occurring in 3% of MEN1 patients, VIPomas in 3%, and GRHomas and somatostatinomas in <1% [39].
Non-functional panNENs are among the most com-mon tumours of the pancreaticoduodenal region in patients with MEN1, with a penetrance as high as 35% at 50 years of age. In most patients, non-functional pan-NENs are ≤2 cm and therefore the risk of metastasis and death is very low [34]. However, average life expectancy for patients with non-functional panNENs is similar to that for patients with gastrinomas and shorter than that for patients without panNENs.
The best way to diagnose and stage non-functional panNENs in patients with MEN1 is unclear. Assays for tumour markers like CgA have low value [40]. Similar to panNENs not associated with MEN1, imaging modalities appear to be ideal for the diagnostic work-up of these patients as well as for the screening of panNENs in patients with MEN1. Current guidelines recommend annual imaging with CT, MRI, or EUS for the screening of panNENs in patients with MEN1 starting at the age of 18 years old [41]. However, given the low growth rate of these lesions, 2-3 years intervals may be considered in patients with previously negative surveillance. Recent data suggest that EUS outperforms CT scanning in this setting, and a combination of MRI plus EUS has been recommended [42]. Furthermore, STTR-PET/CT scanning has been reported to have high sensitivity for panNEN in MEN1, often leading to a change in management [43]. However, high radiation dose has been reported during surveillance program in patients with MEN1 [44]. As such, multi-modality imaging strategies designed to minimize radiation exposure should be considered.
The management of non-functional panNENs in MEN1 is controversial [45]. Many experienced centres have been using a tumour size threshold of 2 cm in the decision to surgically resect non-functional panNEN [46], with subsequent yearly surveillance [47]. Generally speaking, tumours under 1 cm have a low risk for substantial growth and metastasis and avoiding surgery with continued surveillance is reasonable. Available data for tumours between 1 and 2 cm are less clear. Triponez et al. demonstrated that the risk of death was low for patients with panNEN <2 cm and proposed a conservative attitude for these patients in the absence of aggressive features such as rapid progression on imaging studies [48]. In patients with MEN1, panNENs are responsible for 30% of the ZES, with duodenal NENs being responsible for the remainder. Most gastrinomas are well differentiated, with a low proliferative activity (Ki67 index), usually close to 2%. Immunohistochemically, almost all gastrinomas stain for gastrin [49]. Patients with ZES and MEN1 present at an earlier age (mean 32-35 years) than patients with sporadic disease [34]. Of all MEN1/ZES patients, 25% lack a family history of MEN1, supporting the need to suspect of MEN1 in all patients with ZES [34]. In up to 45% of MEN1 patients, the symptoms of ZES precede those of hyperparathyroidism and can be the initial symptoms [50]. However, almost all MEN1 patients have hyperparathyroidism at the time of ZES diagnosis, although in many patients it can be asymptomatic [34, 50]. Regarding insulinomas, approximately 5% are associated with MEN1 syndrome. Comparative studies between patients with insulinomas, with or without MEN1 are lacking. Finally, the association between MEN1 and rare functioning tumours is less clear. Because of the frequent association between MEN1 and ZES, and in similarity to patients with a clinical diagnose of MEN1 (Table 5), all patients with ZES should have biochemical studies for MEN1. Similarly, all patients with insulinoma or rare functioning tumours with suspicion of MEN1 (Table 5) should have the same evaluation. Serum parathormone levels, ionized calcium levels and prolactin levels should be performed at initial evaluation and during yearly follow-up in patients with ZES [34, 50]. Furthermore, all patients suspected of MEN1 (Table 5) need to be assessed for the other tumours, which are generally non-functional [49]. Specific parathyroid studies are required if hyperparathyroidism is found (US, CT/MRI, 99m Tc-sestamibi scan) [51]. All patients require MRI of the sella turcica region and, after 20 years of age, require CT of the chest/abdomen [52]. If MEN1/ZES is present, UGI endoscopy for gastric NEN is recommended [53]. Routine SRS is not recommended if other imaging studies for NEN are negative. EUS is more sensitive than cross sectional imaging studies (CT, MRI, US) for the detection and characterization of small non-functional panNENs. However, since routine surgical resection of small panNENs (<2 cm) is not recommended and the EUS criteria on when to operate these patients are not established the added benefit of EUS is controversial [54].

Regarding genetic testing, it should be performed in patients meeting criteria for MEN1 (≥2 tumours associated with MEN1), patients with gastrinomas or patients meeting further criteria described in Table 5. If genetic testing is considered, genetic counselling should be offered, prior to testing [51, 55].
In MEN1/ZES patients, surgery without a Whipple resection is associated with >90% of recurrence [34, 49, 56]. Therefore, routine surgical exploration is controversial in patients with MEN1/ZES. Indeed, these patients usually have multiple gastrinomas, frequently with lymph node metastases, with concomitant panNENs (non-functional primarily), are rarely cured and have an excellent life expectancy if only small tumours (<2 cm) are present [34, 49, 57]. However, surgery is the only treatment approach with curative intent [56]. As such, it has been generally recommended that surgery should be performed in patients with MEN1 and panNENs >2 cm [48]. In patients with MEN1 and insulinoma, in which multiple tumours are frequently present, the aim of surgery is to control inappropriate insulin secretion by excising all insulinomas. As such, preoperative ocalization of which pancreatic tumours are the insulinomas is mandatory because these patients frequently have other panNENs (usually non-functional) [58].
The prognostic significance of MEN1 in patients with panNENs is not entirely clear. Some studies in patients with gastrinomas suggest these patients have a better prognosis, even though the gastrinomas are almost always multiple [59]. With the ability to treat both the ZES and the hyperparathyroidism, recent studies show that in patients with MEN1, the natural history of the panNEN is increasingly becoming a determinant of survival [34]. Finally, patients with MEN1 frequently have multiple insulinomas, however, these are usually cured surgically [60]. There are no comparative studies on survival in MEN1 patients with insulinomas compared to sporadic cases.
Von Hippel-Lindau Disease
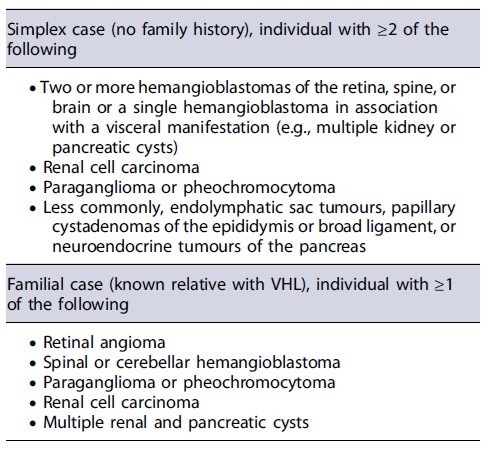
VHL is a rare autosomal-dominant disease caused by mutations in the VHL gene, on chromosome 3p25, that encodes the peptide pVHL, important in the regulation of angiogenic growth and the activity of several mitotic factors (VEGF, PDGF, TGFα, erythropoietin) [34]. VHL is characterized by hemangioblastomas of the retina and cranio-spinal region, endolymphatic sac tumours, renal cell carcinomas or cysts, pheochromocytomas, and epididymal cystadenomas (Table 6). Furthermore, pancreatic tumours or cysts can be present in 35-77% of patients [46, 59]. Specifically, panNENs develop in 10-17% of patients with VHL, and in almost all cases they are non-functional panNENs, and usually are asymptomatic (Table 4) [61]. In contrast to MEN1 patients, most VHL patients have a single panNEN, although patients might also present with multifocal lesions [34]. The majority of panNENs in VHL are well differentiated (grade 1 or 2), small (<2cm) and present a slow growth when compared with sporadic tumours. As a result of its clinical indolence, many of these lesions are diagnosed incidentally during routine VHL surveillance for renal lesions [62]. However, in 8-50% of VHL patients, panNENs are metastatic and LMs occur in 9-37% [63]. In patients with VHL, panNENs are more likely to metastasize when present with size >3cmin diameter, rapid tumour doubling time (<500 days), and VHL missense and/or exon 3 pathogenic variants [64]. Based on these 3 risk factors, risk stratification of panNENs in the context of VHL has been suggested for management optimization [64]. As such, in the presence of a panNEN, if clinical features suggest VHL (Table 6), appropriate gene testing should be considered after genetic counseling [34], and molecular imaging, typically with STTR-PET/CT should be offered [65].
Management of panNENs is primarily surgical, although the criteria for surgical resection differ from those of patients with sporadic panNENs. Surgical resection should be reserved for patients with potentially resectable lesions greater than 3 cm in diameter in the body or tail of the pancreas, or greater than 2 cm in diameter in the head of the pancreas [65]. The hypoxia-inducible factor-2alpha (HIF-2alpha) inhibitor Belzutifan, rather than other systemic therapies, is considered the best approach if surgery is not feasible, or the tumour is considered unresectable [65]. Nonoperative approaches (e.g., surveillance, Belzutifan) are appropriate for small primary lesions (≤3 cm), incorporating other clinical factors such as type and location of the VHL pathogenic variant, and rate of tumour growth [63]. Regarding prognosis, VHL-associated panNENs tend to be associated with a more indolent course and, similarly, long-term outcomes of resected VHL-associated panNENs appear to be generally better than those of sporadic [66].
Neurofibromatosis Type 1
NF-1 is an autosomal-dominant syndrome with a population frequency of 1 in 3,000 births, with half of cases due to a de novo mutation [67]. NF-1 is due to mutations in the NF-1 gene on chromosome 17q11.2, which encodes the protein neurofibromin, which affects cell growth, through Ras protein activation and mammalian target of rapamycin (mTOR) cascade regulation [46, 62]. This condition is characterized by the development of several tumours, including neurofibroma, pheochromocytoma, and GI stromal tumour (Table 1)[46, 62]. CNS abnormalities are frequent with learning disorders (30-60%), attention deficit hyperactivity disorder, and epilepsy [46, 62]. Furthermore, panNENs occur only in a minority of NF-1 patients (10%) and are almost exclusively duodenal somatostatinomas (Table 4)[46, 63, 64]. However, NF-1 patients have been reported with NF-panNENs, ZES and insulinomas [34]. Duodenal somatostatinomas characteristically occur in the peri-ampullary region, have a mean size of 2.8 cm (range 1-5), comprise 23% of all ampullary NENs in several series, metastasize in 30% of cases, and are rarely associated with the clinical somatostatinoma syndrome (1-2%) [46]. To this date, there are no data to allow specific management recommendations of panNENs in the context of NF-1 and patients with NF-1 with panNENs are usually treated as sporadic panNENs. However, it is important to remember that in patients with panNENs, if clinical features suggest NF-1, appropriate gene testing should be considered after genetic counseling [34].
Tuberous Sclerosis Complex
TSC is an autosomal-dominant disease caused by a mutation in one of two genes: the TSC1 gene (which encodes hamartin) or the TSC2 gene (which encodes tuberin). Both hamartin and tuberin are codependent and play a role in mTOR cascade regulation, protein translation, protein synthesis, and cell proliferation [34]. This condition is characterized by the development of hamartomas in multiple organs, neurological features (autism, mental retardation, epilepsy), and dermatological features (hypomelanotic macules, shagreen patches, ungual fibromas, facial angiofibromas) [68]. A small percentage of TSC patients (1%) have been reported to have panNENs, including gastrinomas, insulinomas, and non-functional panNENs, some of which are metastatic (Table 4) [69]. As such, in patients with panNENs, if clinical features suggest TSC, appropriate gene testing should be considered after genetic counselling [34]. However, to this date, specific management recommendations of panNENs in the context of tuberous sclerosis are lacking and patients are usually treated as sporadic ones.
Conclusion
Among pancreatic neoplasms, panNENs are rare tumours. However, their incidence is increasing with the advancement of imaging technology and increased sporadic form of the disease. The unique aspects to management, challenges in hereditary disease recognition and accurate diagnosis, and rarity of these syndromes should be kept present during the evaluation of patients with panNENs.

